Introduction to Integrative Multi-omics Analyses
One of the first stages in microbial systems biology has been allocated to genomics by the convenience at which genomes are presently sequenced. Regardless of the method utilized, genome sequencing is usually accompanied by arrangement and encoding and returns an almost full view of a given microorganism's genetic reservoir. On the other side, the genome sequence is only a summary of an organism's actual phenotypic capacities, offering very little evidence on other key parts of the underlying life cycle, like the reaction to environmental and genetic disturbances, time fluctuations, gene essentiality, etc. Fixed information derived from the genome sequence is not sufficient to obtain a systemic and comprehensive summary of living entities, and other levels of understanding must be taken into account.
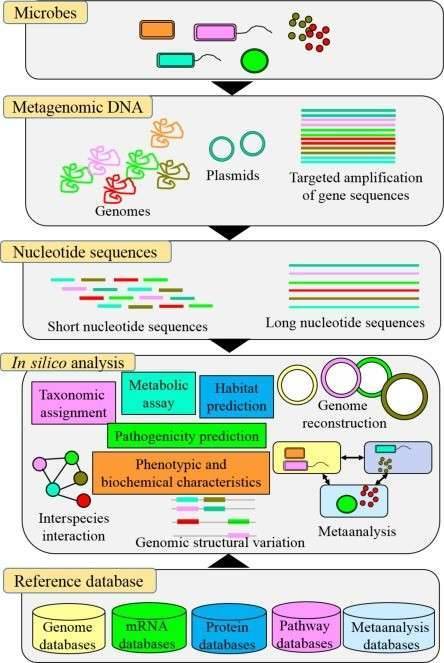
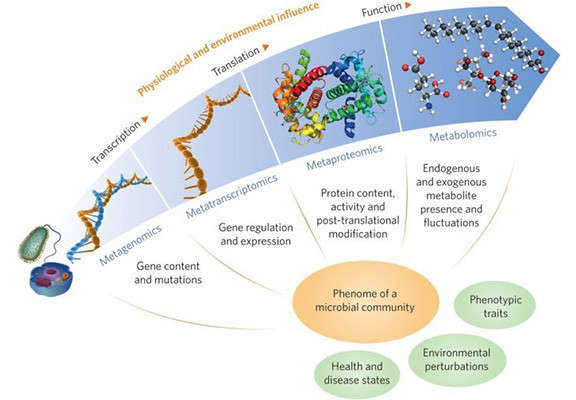 Figure 1. A multi-omic future for microbiome studies (Jansson, 2016)
Figure 1. A multi-omic future for microbiome studies (Jansson, 2016)
Recent advances in numerous 'omics' innovations enable large quantifiable tracking of the enormous amount of different biological molecules and thus enable the identification of their variance on a genomic scale between distinct biochemical states. Numerous famous 'omics' technologies used in the biology of microbial processes involve transcriptomics, which quantifies the rates of mRNA transcript; proteomics, which evaluates the number of proteins; metabolomics, which assesses the number of small cell metabolites; interactomics, which settles the entire collection of cellular molecular interrelationships; and fluxomics, dynamic shifts of molecules within a cell in the course of time. Even so, no single analysis of 'omics' can completely uncover the intricacies of basic microbial biology. In order to obtain an accurate view of living microorganisms, a combination of various pieces of encryption, the multi-'omics' method, is thus needed. Despite this being a difficult mission, some efforts have recently been developed to assimilate heterogeneous 'omics' databases into different microbial platforms and the findings have shown that the multi-'omics' method is a strong device for comprehending the functional ideals and fundamentals of complete cellular systems.
Experimental 'omics' Approaches
Cells are living processes with lots of various functional molecules, which ultimately decide the cells' phenotype. Such molecules involve DNA transcribed mRNA, mRNA translated proteins, and numerous different molecular mass metabolites generated by different enzymatic activities. Clearly, it is not sufficient to analyze the DNA sequences of microbial genomes alone to acquire crucial information on the capabilities of these molecules and the regulatory processes engaged in the making of those molecules. The previous decade has seen substantial growth in methods related to molecular biological assays to quantify different cellular molecules in order to address the limitations in genome analysis, and these attempts have contributed to the emergence of different 'omics' experimental methods.
Transcriptomics
Transcriptomics, also known as global gene expression analysis or profiling of genome-wide expression, is one of the instruments for measuring the entire collection of all mRNA molecules created in one cell or cell population, or 'transcripts'., Transcriptomics has allowed quantitative observations of the diverse expression of mRNA molecules and their variance between distinct states in the genome-scale, as opposed to genome sequencing and comparative genomics innovations that concentrate on DNA, which is fixed data for any provided microbial species and usually does not alter considerably in reaction to short-term external environmental adjustments, thus reflects the genes that, with the exception of mRNA deterioration occurrences, are constantly demonstrated at any given moment.
Proteomics
It pertains to the analysis of the framework, purpose, and abundance of proteins at the level of the whole cell. It enables the quantification of the level of cellular protein expression; it can also shed light on post-translational alterations, the interconnection between protein and protein, and the translation of protein. Proteomics has depended strongly on two key protein segregation and visualization methods: (i) two-dimensional PAGE (2D-PAGE), in which proteins are divided by their isoelectric point and mass, accompanied by mass spectrometric detection, and (ii) Gel-free profiling processes that combine micro-scale phase separation with automated conjunction mass spectrometry, such as multidimensional separations.
Interactomics
Cellular life has been proposed to be coordinated through a vast system of protein interrelationship, in which many proteins engaging in the same operational paths appear to accumulate in multi-component protein compounds together. Thereby, the large-scale identification of these collated proteins, namely the interactomics, is one of the key paths of operational proteomics and could offer a new perspective into the biosynthesis of microbial cells.
Metabolomics and Fluxomics
Metabolomics illustrates many benefits in terms of metabolic assessments as a way to describe the small-molecule diversity in the cell and to display distinctions in the profusion of small molecules because metabolites are the operational individuals within the cells and their concentration rates differ as a result of genetic or physiological shifts. In addition, microbial metabolomics research using isotope-labeled intermediate metabolites coupled with different kinds of dynamic metabolic flux modeling has been proposed as a valuable tool in research into intricate and large-scale metabolic processes in some latest applications, and the term 'fluxomics 'has been created to define this course of research.
Epigenomics
At single-base and strand resolution, epigenomics can now be evaluated and plays a crucial role because it can occasionally illustrate phenotypic distinctions between cells with extremely similar genetic details.
Phenomics
Phenomics (high-throughput phenotyping) enables automated and large-scale exploration of the phenotypic space of a provided species and the derivation of phenotype capacities (e.g. the tendency to metabolize specific carbon sources with respect to others).
References
- Lloyd-Price J, Arze C, Ananthakrishnan AN, et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature. 2019 May;569(7758).
- Jansson JK, Baker ES. A multi-omic future for microbiome studies. Nature microbiology. 2016 Apr 26;1(5).
- Fondi M, Liò P. Multi-omics and metabolic modelling pipelines: challenges and tools for systems microbiology. Microbiological research. 2015 Feb 1;171.