
 Sample Submission Guidelines
Sample Submission Guidelines
A Guide of Single-Cell DNA Methylation Sequencing: From Wet Experiment to Data Analysis
What is single-cell DNA methylation?
DNA methylation is an epigenetic marker inherited during cell division that affects the biological function of cells. Differences between cells may correspond to differences in epigenetic markers, which in turn are related to gene expression. In turn, genome-wide methylation analysis at the single-cell level will provide insight into transcriptional regulation and cellular heterogeneity. Exploring epigenetic differences between cells is key to understanding tissue heterogeneity. Recent advances in single-cell DNA methylation mapping provide an opportunity for the discovery of such heterogeneity.
How to do single-cell DNA methylation profiling?
Bisulfite Conversion Methodology
The bisulfite conversion method is considered to be the gold standard for DNA methylation analysis. It is favored by researchers due to its high conversion rate (>99%), reproducibility and simplicity of use via commercial kits.
The main differences between RRBS and WGBS
RRBS and WGBS are popular methods for genome-wide methylation analysis. Both methods include bisulfite conversion and NGS preparation. The main difference is that RRBS uses appropriate restriction endonucleases and size selection to screen for GC-rich regions. GBS (especially MethylC-seq) has the advantage of being able to cover most of the CpGs in the genome. the purification and screening process is relatively simple with WGBS compared to RRBS. Preventing degradation losses during bisulfite transformation in WGBS is considered relatively important and therefore many single-cell methods based on WGBS are often based on PBAT.
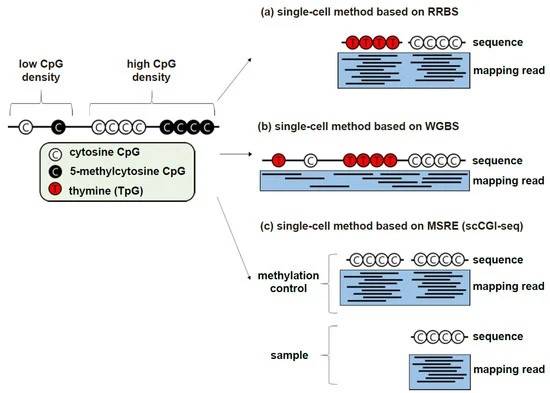 scRRBS and scWGBS (Ahn J et al., 2021)
scRRBS and scWGBS (Ahn J et al., 2021)
The workflow of single-cell RRBS
Conventional RRBS can be divided into five steps. The first step is the digestion of DNA with restriction endonucleases, the second step is splice ligation and its pre-treatment, the third step is the selection of GC-rich sites, the fourth step is bisulfite transformation and finally the amplification process prior to sequencing. These processes include several purification steps, with a final amplification step that is designed to compensate for losses in previous processes. Unlike conventional RRBS, single-cell RRBS does not guarantee that the target DNA will remain intact during the final amplification process due to the small amount of DNA input from a single cell by conventional methods. Therefore, single-cell RRBS is refined in terms of loss reduction. scRRBS is a method that focuses on minimizing the losses that occur during the purification process. Loss reduction is achieved through restriction endonuclease digestion, the splice ligation process and transformation of bisulfites in a single tube without purification.
The workflow of single-cell WGAS
WGBS is whole genome methylation sequencing and provides more comprehensive genomic coverage than the RRBS method which covers only CpG islands. WGBS is simpler than RRBS as it does not require enzyme digestion, size selection and sample maceration and purification steps. However, the loss of sample in WGBS is mainly due to bisulfite conversion. It can also be optimised by the PBAT method.
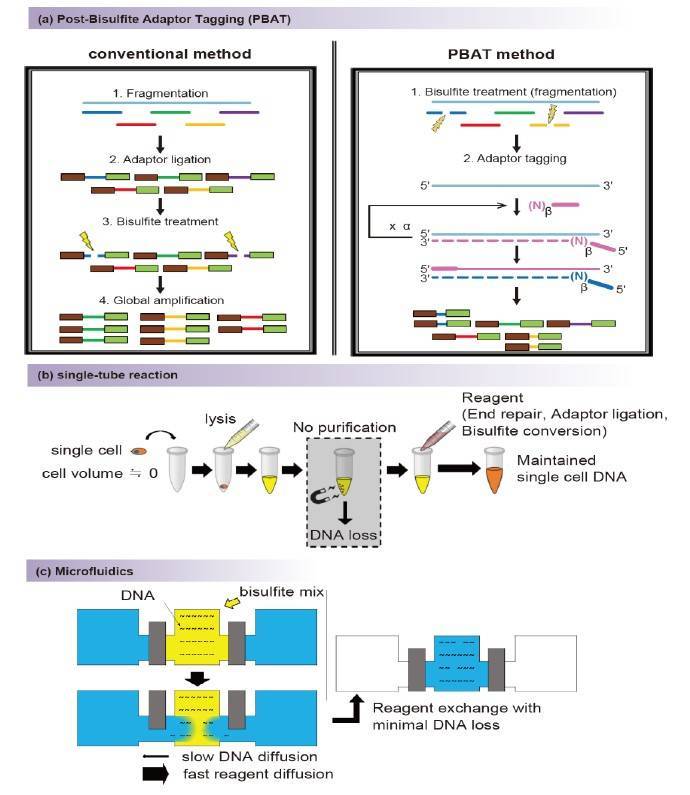 (a) Overview of the post-bisulfite adaptor tagging (PBAT) method to prevent loss due to degradation during the bisulfite conversion process. (b) Overview of single-tube reaction. (Ahn J et al., 2021)
(a) Overview of the post-bisulfite adaptor tagging (PBAT) method to prevent loss due to degradation during the bisulfite conversion process. (b) Overview of single-tube reaction. (Ahn J et al., 2021)
BS Conversion-Free Methodology
Methods for methylation sequencing without BS conversion fall into two main categories: those that exploit the affinity binding of methylcytosine and those that exploit the sensitivity of restriction endonucleases to methylcytosine. MBD-seq and MeDIP-seq are representative methods with affinity-based profiles. Affinity-based methods are not suitable for application on single-cell sequencing because they are based on DNA fragments to produce an average DNA methylation profile and therefore cannot distinguish differences in DNA methylation patterns in individual cells. However, unlike affinity-based methods, MSRE-based methods have been refined for use in single-cell sequencing, and scCGI-seq measures methylation in a similar way to Methyl-seq.
Currently, the bisulfite conversion method remains one of the most common methylation assays. Therefore, the next presentation on analytical methods uses the bisulfite conversion method as an example.
Single-cell DNA methylation sequencing data analysis
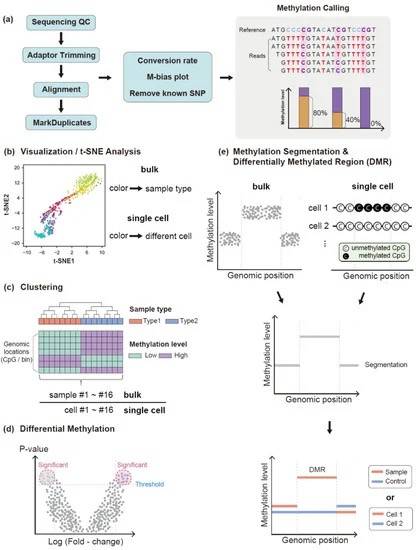 Analysis methods of DNA methylation. (Ahn J et al., 2021)
Analysis methods of DNA methylation. (Ahn J et al., 2021)
Data quality assessment
After sequencing experiments including RRBS and WGBS have been performed, the sequence data needs to be pre-processed. The pre-processing steps can be divided into data quality control (QC), trimming of sequencing reads and comparison of sequencing reads. The first part of QC measures the overall basic sequencing data quality using software programs such as FastQC. Following a thorough sequence quality assessment, the conversion efficiency of bisulfite sequencing should be determined. For samples with low heavy sulfite conversion rates, it is challenging to distinguish between bases sequenced as cytosines at the CpG site and true methylated cytosines or sequencing errors. This plays a role as a confounding factor in downstream analysis, including finding differentially methylated regions. Conversion efficiency can be obtained directly from the added unmethylated lambda DNA by calculating the proportion of converted bases at the cytosine position or by using available software programs.
Sequence clipping and comparison
Sequence clipping is performed using the software and the trimmed sequence reads are then compared to the reference genome, which can detect and remove low-quality or ambiguous base calls, as well as adapter sequences that may have been introduced during library preparation.
Methylation analysis using methylation level
The main aim of methylation analysis is to explore epigenetic evidence of differences between the constituent samples, organs and disease states (including cancer). In order to discover these differences, a numerical value characterizing this concept is required, commonly a beta value, which is calculated using the formula below. After the methylation call, subsequent analyses such as visual analysis of t-SNE, cluster analysis, and identification of differentially methylated cytosines (DMCs) or differentially methylated regions (DMRs) are performed.
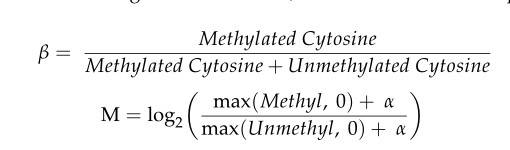
Methylation analysis using Reads methylation patterns
Recent methylation analyses have utilized the methylation patterns of each read to diagnose diseases, particularly cancer. This new analytical concept is based on the biological property of methylation, i.e., the tendency to maintain methylation between adjacent CpG sites unless ab initio methylation occurs. This reads pattern analysis method is able to detect DNA molecules with disease signals and has the potential to increase the chances of disease signal detection. For example, a large liquid biopsy study designed an integrated classifier to classify tumor types based on reads pattern analysis and showed significant results in the detection of early-stage cancers. In addition, the quantification of tumor-derived DNA molecules by methylation patterns is another way to look at tumor burden.
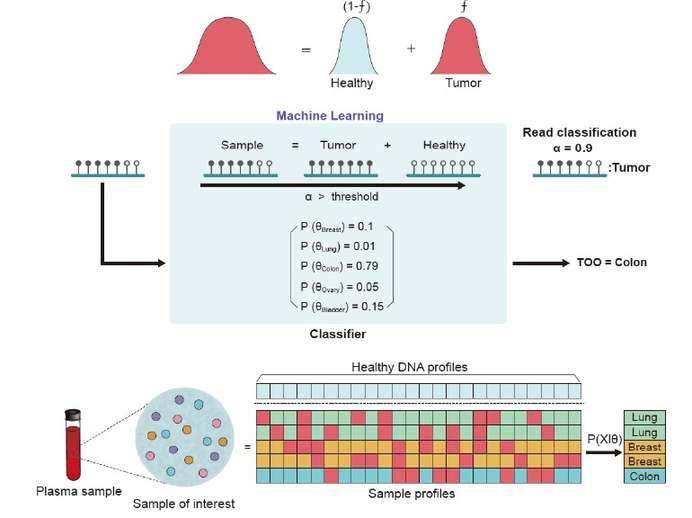 Methylation analysis using Reads methylation patterns. (Ahn J et al., 2021)
Methylation analysis using Reads methylation patterns. (Ahn J et al., 2021)
Applications of Single-cell DNA methylation sequencing
Cell development research
The maturation of germ cells or embryonic cells is influenced by the expression of specific genes, which correlate with the level of methylation in DNA. Based on the methylation profile of pre-implantation embryonic cells, for example, the mechanisms of pre-implantation cell methylation and its phenomena have been investigated using single-cell methylation sequencing by tracking early embryonic lines. The team observed that non-CpG methylation accumulates during oocyte maturation, suggesting a different role for non-CpG methylation than for CpG methylation during oocyte maturation.
Disease-related research
In patients with the disease, the pattern of DNA methylation differs from that of healthy individuals. Among the various diseases, cancer in particular has a pattern of DNA methylation that normal cells do not have, leading to differences in gene expression levels. In the study of cancers with this heterogeneity, a multi-omics approach combining genomic variation and RNA expression needs to be used for analysis. For example, a research group has recently developed a method called scTrio-seq2, which integrates single-cell transcriptome and single-cell methylation sequencing data. Several studies have shown that a multi-omics approach using single-cell methylation sequencing (sc-methyl-seq) can overcome the limitations of previous methods and provide better discrimination. Thus, sc-methyl-seq can be used in various fields to address fundamental questions related to biological processes and diseases.
Reference
- Ahn J, Heo S, Lee J, et al. Introduction to single-cell DNA methylation profiling methods[J]. Biomolecules, 2021, 11(7): 1013.


