
 Sample Submission Guidelines
Sample Submission Guidelines
Whole Virus Genome Sequencing in Screening New Drugs
Whole genome sequencing of viruses is playing an increasingly important role in clinical practice. On the one hand, analyzing the drug resistance information of the virus can assist in more accurate clinical diagnosis, treatment, and disease course management; On the other hand, through the establishment of molecular epidemiology of the virus, strict control of viral infection can be achieved. Based on high-throughput sequencing technology, the isolated virus was sequenced and analyzed to obtain all the genome information of the virus, and the virus virulence system, genome evolution and evolution process were explored through structural genomics and comparative genomics at multiple levels, for a better understand the diversity, ecology, adaptability, and evolution of viruses.
Virus Genome Sequencing Helps Scientific Drug Use and Vaccine Development
The SARS-CoV-2 virus as an RNA virus, the causative agent of COVID-19, is undergoing constant mutation. There is a possibility that mutations accumulated during a pandemic could produce measurable effects on the infected population or complicate epidemic control efforts, highlighting a need to monitor the genetic diversity and epidemiology of SARS-CoV-2 throughout the pandemic. Researchers collaborated with 20 hospitals and institutes and analyzed epidemiological, genetic, and clinical data collected in Sichuan province of China across the outbreak period from 22 January to 20 February 2020. The research team also used the nanopore sequencing technology to sequence the whole genome of the virus in 310 clinical samples from 248 COVID-19 infected people, and verified it with sequencing technology. The results showed that 35 recurrent variants were identified and about 20% of them have the same mutant Nsp1 coding region( Δ 500-532) missing. Subsequently, the molecular epidemiology of the mutant was analyzed. The public virus database showed that the Nsp1 deletion mutant had appeared in the virus database of 37 countries and regions. Based on 117 clinical data, the study found that this mutant strain is associated with lower viral load levels and lower peripheral blood IFN- β Horizontally related; And Nsp1 was confirmed through experiments in different cell lines The Δ500 532 mutant strain can reduce the activity of the IFNB1 promoter and further inhibit downstream IFN-I signaling. This study provides clues for the use of these genomic markers in molecular epidemiology investigations, and potentially important information for the effective design of vaccines and drugs for COVID-19.
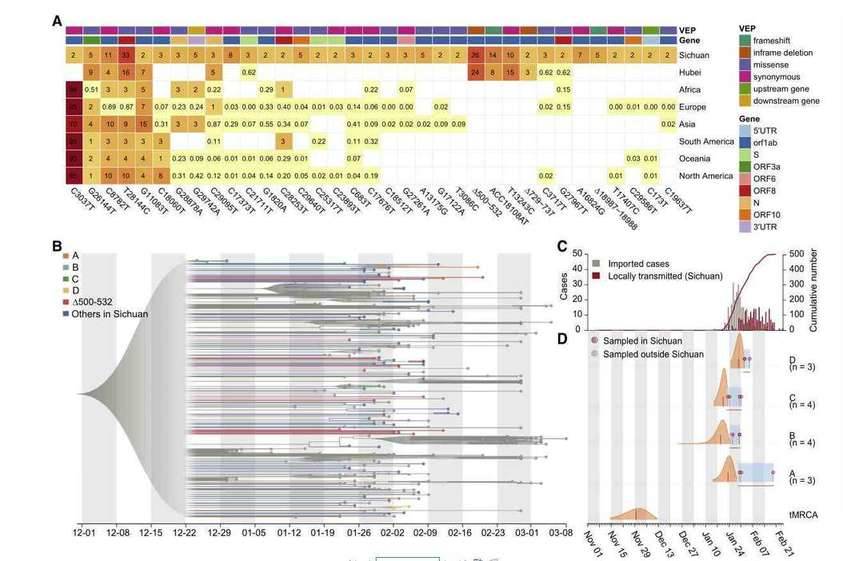 Recurrent genetic variants and phylogenetic analysis in the SARS-CoV-2 genomes
Recurrent genetic variants and phylogenetic analysis in the SARS-CoV-2 genomes
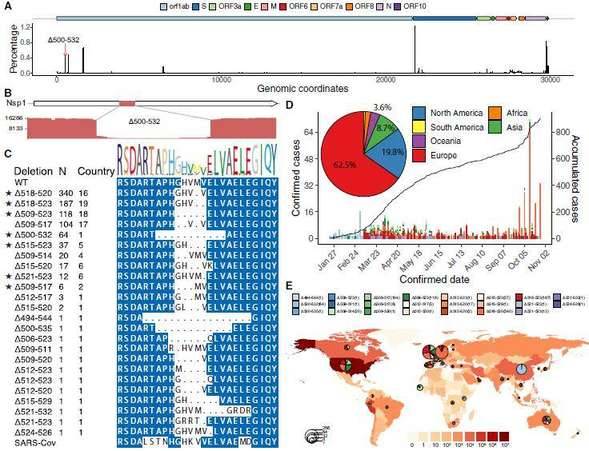 Prevalence of deletion variants in the 500-532 locus in SARS-CoV-2 genome
Prevalence of deletion variants in the 500-532 locus in SARS-CoV-2 genome
Virus Genome Sequencing for Discovering Novel Antibiotics
Enterohemorrhagic Escherichia coli (EHEC) O157:H7 and Enterotoxigenic E. coli (ETEC) are important foodborne pathogens, causing serious food poisoning outbreaks worldwide. As a new type of antibacterial agent through genome and phylogenetic analysis, bacteriophages are increasingly being used to control foodborne pathogens. Researchers have studied a novel wide host bacteriophage vB through whole genome sequencing_ EcoM_ SQ17 (SQ17), which has the ability to control the number of bacteria in food. The genome of bacteriophage SQ17 is a linear double stranded DNA of 166457 bp, with a GC content of 32.52%. The genome contains 258 putative ORFs, of which 136 are classified as putative proteins. These proteins aggregate into four functional groups related to tail assembly, capsid assembly, host lysis and lysis inhibition, replication, transcription, and repair. Genomic analysis showed that phage SQ17 had no genes related to antibiotic resistance, toxin, lysogenicity or virulence factor, and was an excellent candidate for potential food applications. SQ17 has a wide host range and can infect EHEC O157: H7, ETEC and other Escherichia coli strains. Result has shown that bacteriophage SQ17 is applied to reduce EHEC O157: H7 and ETEC pollution in fresh lettuce, and SQ17 has high infectivity and rapid replication rate, making it more suitable for application in the food industry. Higher temperatures (25 ℃) can affect the bactericidal effect of bacteriophages, therefore, it is not recommended to store or process fresh food at high temperatures for a long time in the food industry.
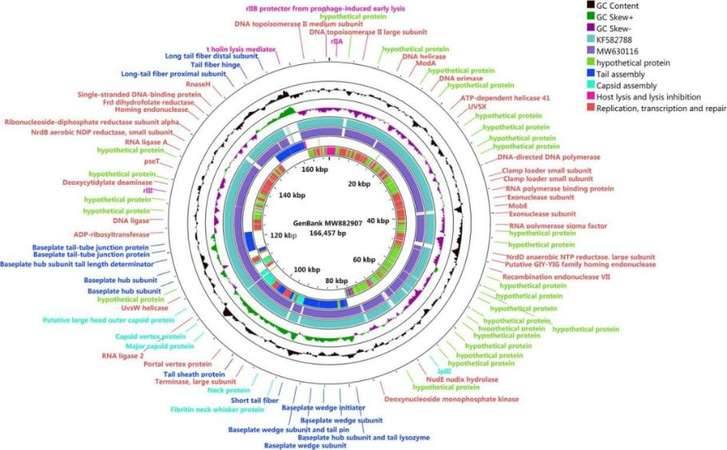 Genomic map of bacteriophage SQ17
Genomic map of bacteriophage SQ17
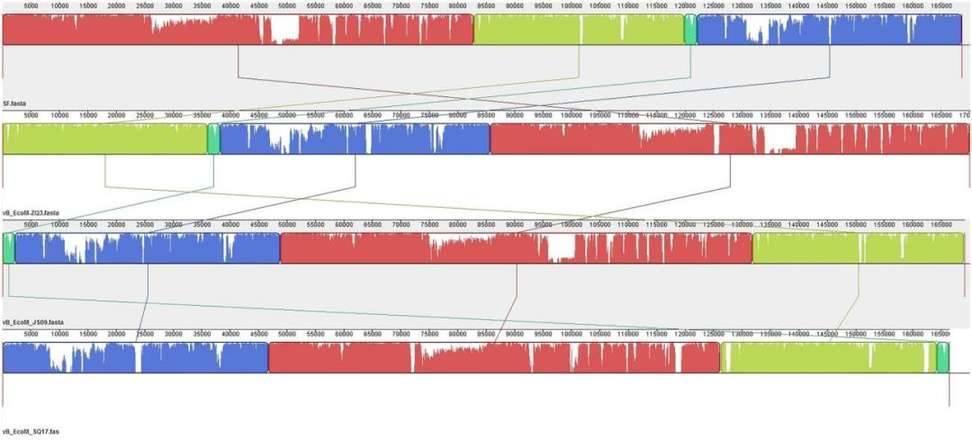 Phage SQ17 and Phage SF, vB_ EcoM-ZQ3 and vB_ EcoM_ JS09 for genomic comparative analysis
Phage SQ17 and Phage SF, vB_ EcoM-ZQ3 and vB_ EcoM_ JS09 for genomic comparative analysis
Whole genome sequencing technologyhas been widely applied in virus mutation monitoring, typing and tracing, especially in monitoring the dynamic changes of virus gene sequences in samples, which has scientific guiding significance for epidemic monitoring. It will be widely used in the future.
References:
- Lin, Jing-Wen et al. "Genomic monitoring of SARS-CoV-2 uncovers an Nsp1 deletion variant that modulates type I interferon response." Cell host & microbe vol. 29,3 (2021): 489-502.e8.
- Zhou, Yan et al. "Application of a novel lytic phage vB_EcoM_SQ17 for the biocontrol of Enterohemorrhagic Escherichia coli O157:H7 and Enterotoxigenic E. coli in food matrices." Frontiers in microbiology vol. 13 929005. 5 Aug. 2022.


