
 Sample Submission Guidelines
Sample Submission Guidelines
HiFi Long-read Sequencing Generates Comprehensive WGS Data to Detect All Types of de novo Mutations
Introduction
Comprehensive characterization of human genomic variants is important for gaining insight into genetic traits and diseases. For rare disease studies, it is particularly important to identify complete maps of all variant types, including indels, short tandem repeats (STRs), and structural variants (SVs). A particular challenge to the accuracy of genome sequencing is de novo mutation (DNM), which has been shown to be a major cause of sporadic, severe, early-onset disease. Each human genome contains on average about 40-90 DNMs, but they are also among the most difficult variants to identify. Therefore, comprehensive detection of all types of DNMs requires high-quality sequencing data.
While short-read sequencing can accurately detect small variants, it has limited sensitivity for large STRs, CNVs and SVs. Long-read sequencing has been widely used for de novo assembly of the human genome and for SV not detected by short-read sequencing, but its low accuracy at single base pair (bp) resolution prevents them from reliably detecting variants smaller than 50 bp. To meet the dual need for long read length and high accuracy in genome analysis, PacBio has developed HiFi long-read sequencing technology that combines both long read length and high accuracy, making long-read sequencing also suitable for detecting small variants.
HiFi Long-read Sequencing Generates Comprehensive WGS Data
A research article titled "Comprehensive de novo mutation discovery with HiFi long-read sequencing" was published in Genome Medicine. The team constructed the most comprehensive mutation dataset with only a single technique, HiFi long-read sequencing, enabling accurate substitution, indel, STR and SV detection. The accuracy of the technique even allows sensitive detection of DNM at all different mutation levels and also allows phasing, which helps to distinguish true-positive from false-positive DNM.
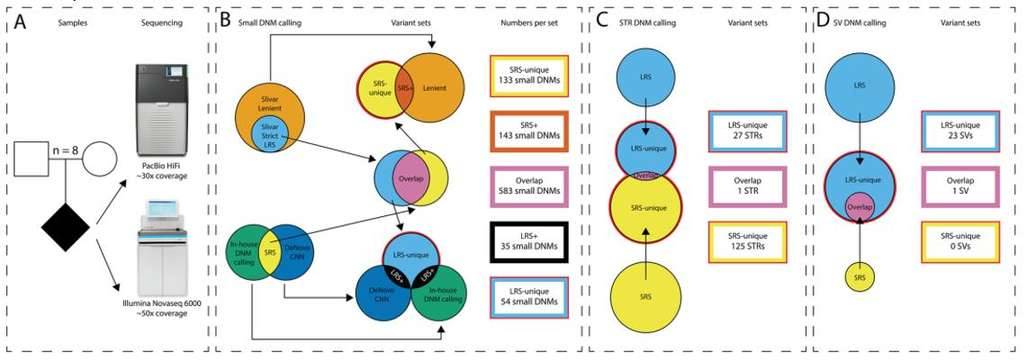 Comprehensive de novo mutation discovery with HiFi long-read sequencing. (Kucuk et al., 2023)
Comprehensive de novo mutation discovery with HiFi long-read sequencing. (Kucuk et al., 2023)
The increased base precision of HiFi sequencing is particularly beneficial for the detection of small DNMs compared to short-read sequencing, and can even improve sensitivity. In addition, unlike other long-read sequencing technologies, HiFi technology does not have highly unstable chemistry, its data quality is more stable, and HiFi sequencing can achieve 99.9% accuracy. HiFi can therefore be a powerful tool for complex challenges in genomics research.
To demonstrate the utility of long-read sequencing in de novo mutation detection, the team sequenced the genomes of eight parent-offspring trios using high coverage depth PacBio HiFi long-read sequencing (~30x) and Illumina short-read whole-genome sequencing (~50x). For HiFi long-read sequencing, the average length of reads obtained from the study was 17 kb. Over 99.0% of the 5.7 million reads per sample aligned to the reference genome, with a mapping quality of 46.5.
The data showed that, on average, 3.8 million substitution mutations per sample were shared between the two sequencing platforms, corresponding to a 94.0% concordance between long-read long and short-read sequencing assays. According to the findings, HiFi long-read sequencing offered around 240 Mb of sequence coverage for the genome, unlike short-read sequencing. Moreover, the Mendelian genetic error rate for HiFi long-read sequencing was only 2.1%, indicating that the majority of variant calls were accurate.
For indels, HiFi long-read long sequencing yielded an average of 1 million mutations per sample and short-read sequencing yielded an average of 900,000 indels. The concordance was 63.1% for short-read sequencing and 58.0% for HiFi long-read sequencing, with approximately 25% of indels being detected in regions not covered by short-read sequencing. In addition, the MIE ratio of indels unique to HiFi long-read sequencing (8.9%) was lower than that of indels unique to short-read sequencing (13.0%), indicating that HiFi long-read sequencing is better at detecting indels.
Overall, the accuracy of HiFi long-read sequencing enables sensitive identification of DNMs at the level of all variants, and also enables fixation, which helps to distinguish true-positive from false-positive DNMs.
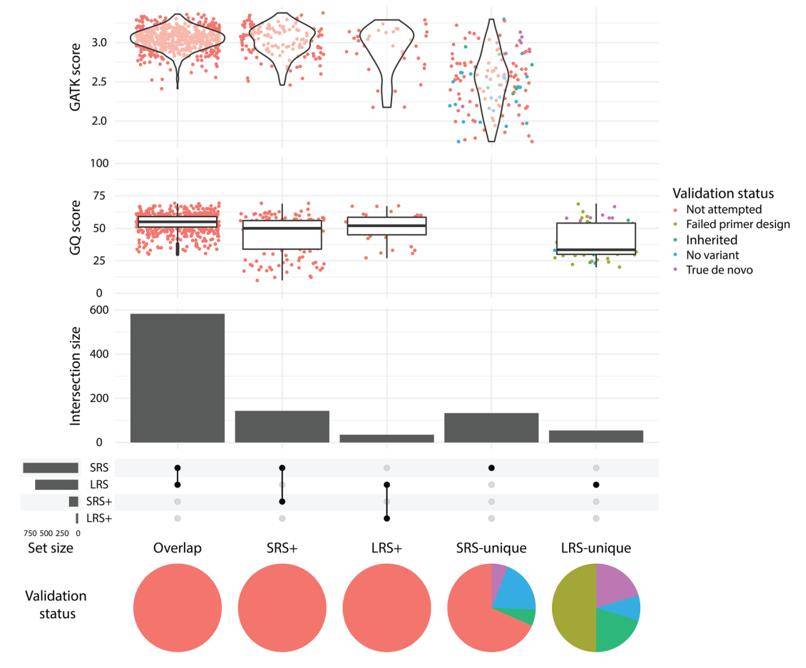 de novo mutation (DNM). (Kucuk et al., 2023)
de novo mutation (DNM). (Kucuk et al., 2023)
A comprehensive WGS dataset can be generated by a single technology, HiFi long-read sequencing, allowing accurate identification of substitutions, indels, STRs and SVs. This means that patients with rare diseases of suspected genetic origin can be tested truly comprehensively with just one comprehensive test. The accuracy of this technology is significant for the diagnosis of severe early onset diseases.
Reference:
- Kucuk, Erdi, et al. "Comprehensive de novo mutation discovery with HiFi long-read sequencing." Genome Medicine 15.1 (2023): 1-15.


