
 Sample Submission Guidelines
Sample Submission Guidelines
ATAC-seq Helps to Analyze Gene Regulation Mechanism
With the deepening understanding of gene regulation mechanism, changes in epigenetics have been found to play an important role. At present, ATAC-seq sequencing technology is an effective method to measure the change of chromatin structure from low level to high level in epigenetics. ATAC-seq technology, which uses Tn5 transposase to sequence protein-free binding regions of the genome, can be combined with chromatin immunoprecipitation coupled with deep sequencing (ChIP-seq) and ribonucleic acid sequencing (RNA-seq) to provide a detailed description of gene expression.
Advantages and application of ATAC-seq
Compared with other technologies, ATAC-seq has the advantages of requiring fewer samples, a shorter sample preparation time and higher reliability and can simultaneously reveal the genome position of open chromatin, the interaction between DNA-binding protein and transcription-binding site. Consequently, it can be used in the following aspects: ① Preliminary judgment on whether the research topic involves epigenetic regulation mechanism; ② Combined with motif analysis, identify transcription factors involved in gene regulation; ③ Identify transcription factors related to cell fate, disease or response and target genes and functional elements regulated by transcription factors; ④ Joint analysis with super enhancer identification to clarify the scope of active super enhancer; ⑤ Understand the gene regulation and cell response mechanism of drugs or diseases.
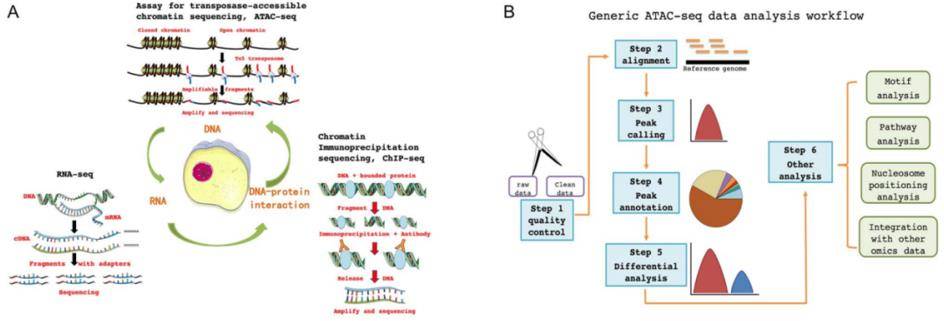 Principles and workflow of ATAC-seq
Principles and workflow of ATAC-seq
Exploration of gene regulatory factors by scATAC-seq
One characteristic of the activity of DNA regulatory elements is chromatin accessibility. However, only active regulatory elements can be recognized and expressed by the cellular mechanism, which shows that chromatin accessibility is closely related to transcriptional regulation. ATAC-seq is applicable to low-cell-number samples, and even single cells, which has enabled epigenomic profiling of primary samples with newfound precision. To date, scATAC-seq has been used to map cell-to-cell variability and rare cell phenotypes, including in healthy and malignant immune cells. The team applies scATAC-seq to obtain chromatin profiles of more than 200,000 single cells in human blood and basal cell carcinoma. This study describes the epigenetic regulation at the single cell level to characterize the development of human immune cells and the regulation of tumor-infiltrating lymphocytes. Additionally, it also illustrates how single-cell ATAC-seq can find cell types and DNA regulatory factors in healthy and diseased tissues.
Examples in human diseases of ATAC-seq
Epigenetic alterations have been considered to be one of the causes of cancer. Cancer studies including pancreatic cancer, liver cancer and bladder cancer, which used ATAC-seq and other sequencing techniques such as RNA-seq and ChIP-seq, have discovered novel regulatory mechanisms and pointed out that the regulation of epigenetics might be a promising anticancer target. Human pluripotent stem cell differentiation towards hematopoietic progenitor cell can serve as an in vitro model for human embryonic hematopoiesis, but the dynamic change of epigenome and transcriptome remains elusive. Here, we systematically profile the chromatin accessibility, H3K4me3 and H3K27me3 modifications, and the transcriptome of intermediate progenitors during hematopoietic progenitor cell differentiation in vitro. Meanwhile, they also used single-cell RNA-seq (scRNA-seq) and single-cell ATAC-seq (scATAC-seq) to uncover the transcriptome and open chromatin features of subpopulations within ECs and HPCs during the EHT window. Comprehensive analyses depict the temporal changes in chromatin configuration and gene expression during hPSC to HPC differentiation and enable the identification of regulators that orchestrate HPC generation. This study describes an epigenome roadmap from human pluripotent stem cells to hematopoietic progenitor cells, which may provide a basis for exploring hematopoietic progenitor cells with better development potential.
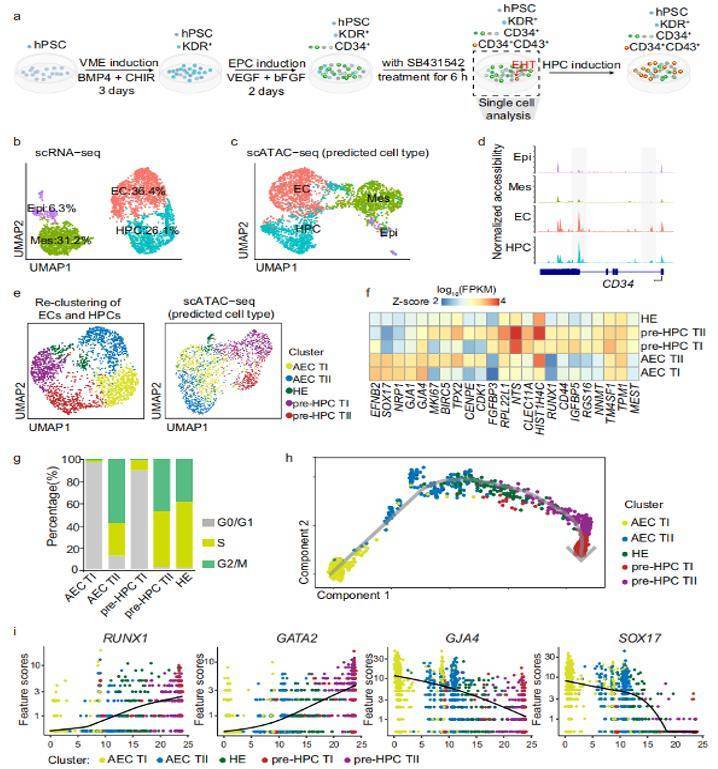 ScRNA-seq and scATAC-seq analysis revealed subpopulations of ECs and early HPCs
ScRNA-seq and scATAC-seq analysis revealed subpopulations of ECs and early HPCs
The emergence of ATAC-seq provides a new perspective for understanding biological mechanisms in terms of chromatin accessibility. It can build different models related to various omics methods according to actual problems and combine them with different technologies. In future scientific research, we firmly believe that it will shine brightly and help us explore more biological mysteries.
References:
- Luo, Liheng et al. "Bibliometric review of ATAC-Seq and its application in gene expression." Briefings in bioinformatics vol. 23,3 (2022): bbac061.
- Satpathy AT, Granja JM, Yost KE, et al. Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion. Nat Biotechnol. 2019;37(8):925-936.
- Chen X, Wang P, Qiu H, et al. Integrative epigenomic and transcriptomic analysis reveals the requirement of JUNB for hematopoietic fate induction. Nat Commun. 2022;13(1):3131. Published 2022 Jun 6.
- Jones, Peter A et al. "Targeting the cancer epigenome for therapy." Nature reviews. Genetics vol. 17,10 (2016): 630-41.


