
 Sample Submission Guidelines
Sample Submission Guidelines
Reduced Representation Bisulfite Sequencing
CD Genomics is offering reduced representation bisulfite sequencing (RRBS) service at single-nucleotide resolution by reducing the amount of sequencing required while capturing the majority of promoters and other relevant genomic regions. RRBS is favorable for biomarker discovery by the identification and analysis of differentially methylated regions (DMRs) between samples.
The Introduction of RRBS
DNA methylation of cytosine is an epigenetic modification involved in gene expression regulation. DNA methylation occurs predominantly in CpG contexts, and these CpG di-nucleotides are more abundant in selected regions of the genome. RRBS is a powerful approach to genome-wide DNA methylation analysis that combines restriction enzyme digestion with bisulfite sequencing to enrich a CpG-dense fraction of the genome. This combination makes RRBS improve the efficiency of the utilization of samples and the perfect platform for pilot studies and clinical applications.
RRBS only sequences the fragments pertaining to CpG-rich regions at a significantly reduced cost when compared with whole genome bisulfite sequencing (WGBS). RRBS is very cost-effective, given that about 1-5% of the genome is sequencing, covering about 12% of genome-wide CpG sites and about 84% of CpG islands in promoters. Plants exhibit a different CpG distribution: (i) lacking characteristic CpG islands in promoters; (ii) occurring at cytosine bases within all sequence contexts. Therefore, while enzyme MspI is commonly used in animals and human, enzyme SacI/MseI is often used in plants.
Advantages of RRBS
- Low input DNA requirement
- Methylation pattern of CpG rich region across the genome
- Covering up to 5 million CpG sites in Human
- Simultaneously detection of DNA methylation and SNPs.
- Epigenetic biomarker discovery
- Single-nucleotide resolution and cost-efficiency
Applications of RRBS
Reduced representation bisulfite sequencing can be used for but not limited to the following research:
Cancer Research: Research into cancer utilizes the RRBS technology to meticulously map DNA methylation profiles across different types of cancers. By scrutinizing the disparities in methylation patterns between cancerous and healthy tissues, RRBS helps pinpoint cancer-specific methylation markers. This sheds light on the epigenetic processes involved in the onset and development of cancer. Additionally, RRBS uncovers specific alterations in DNA methylation that could potentially be utilized as early indicators for innovative diagnostic strategies. This approach underscores the pivotal role of epigenetics in cancer research and emphasizes the pursuit of novel biomarkers for early detection and intervention.
Developmental Biology: RRBS technology is a valuable asset for investigating the multifaceted changes in DNA methylation that occur during embryonic development. It permits a comprehensive analysis of stage-specific and cell-type-specific methylation patterns, shedding light on the intricate dynamics of epigenetic regulation. Furthermore, RRBS enables the observation of DNA methylation modifications throughout stem cell differentiation, unraveling the epigenetic mechanisms that intricately orchestrate cellular fate determination and gene expression.
Neuroscience: In neuroscience, RRBS investigates DNA methylation profiles in neurological disorders such as Alzheimer's disease and autism, uncovering their epigenetic basis and enhancing diagnostic and therapeutic strategies. Furthermore, RRBS explores DNA methylation changes linked to memory formation and learning processes, advancing understanding of epigenetic regulation in neural function and behavior.
Evolutionary and Ecological Studies: RRBS technology serves as a powerful tool for conducting comparative studies on DNA methylation patterns among diverse species or populations in the realm of evolutionary and ecological research. Through this methodology, researchers can delve into the impacts of epigenetics on evolutionary dynamics and adaptive responses. Furthermore, RRBS delves into the repercussions of environmental factors, such as pollution and nutritional variations, on the intricate modulation of DNA methylation, thus uncovering the intricate interplay between epigenetic alterations and environmental influences.
Agricultural Science: In the realm of agricultural science, RRBS serves as a pivotal tool for scrutinizing DNA methylation profiles within crop systems pertinent to crucial attributes including disease resistance, yield capacity, and product quality. Such analyses provide critical guidance for breeding initiatives aimed at advancing the qualities of crop varieties. Moreover, in the arena of livestock breeding, RRBS discerns epigenetic indicators associated with sought-after production traits and general well-being, thereby contributing insights that inform strategic breeding choices and elevate the efficacy of breeding regimes.
RRBS Workflow
The general workflow for RRBS sequencing is outlined below. RRBS utilizes the MspI restriction enzyme to cut at CCGG sites across the genome, enriching CpG-dense regions. The regions containing CpGs are then recovered by gel purification. After methylated adaptor ligation, end repair and dA tailing, theses fragments are then converted by bisulfite, amplified, and sequenced. Our highly experienced expert team executes quality management, following every procedure to ensure confident and unbiased results.

Service Specification
Sample Requirements
|
|
 Click |
Sequencing Strategies
|
 |
Data Analysis We provide multiple customized bioinformatics analyses:
|
Analysis Pipeline
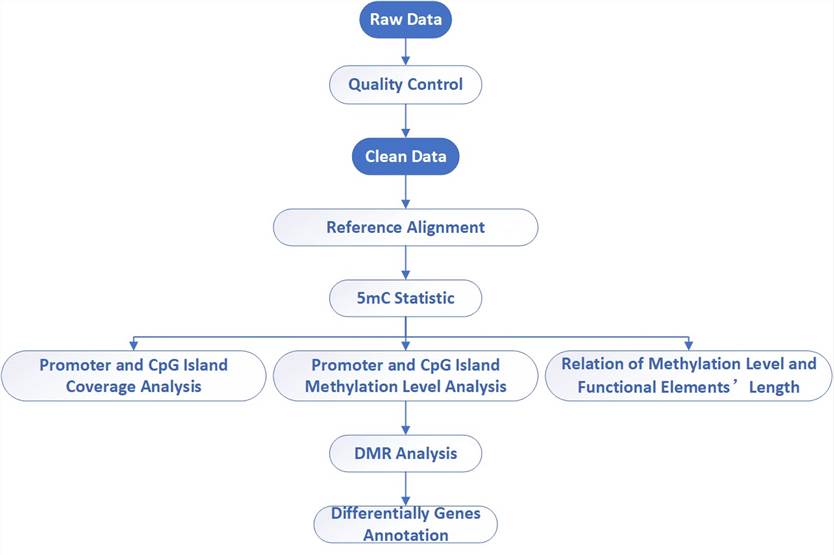
Deliverables
- The original sequencing data
- Experimental results
- Data analysis report
- Details in reduced representation bisulfite sequencing for your writing (customization)
By combining enzyme digestion, bisulfite conversion and NGS technology, CD Genomics is able to provide RRBS as an alternative way to generate and profile genome-wide methylomes at a reduced cost. If you have additional requirements or questions, please feel free to contact us.
References:
- Guo H, Zhu P, Guo F, et al. Profiling DNA methylome landscapes of mammalian cells with single-cell reduced-representation bisulfite sequencing. Nature protocols, 2015, 10(5): 645.
- Meissner A, Gnirke A, Bell G W, et al. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic acids research, 2005, 33(18): 5868-5877.
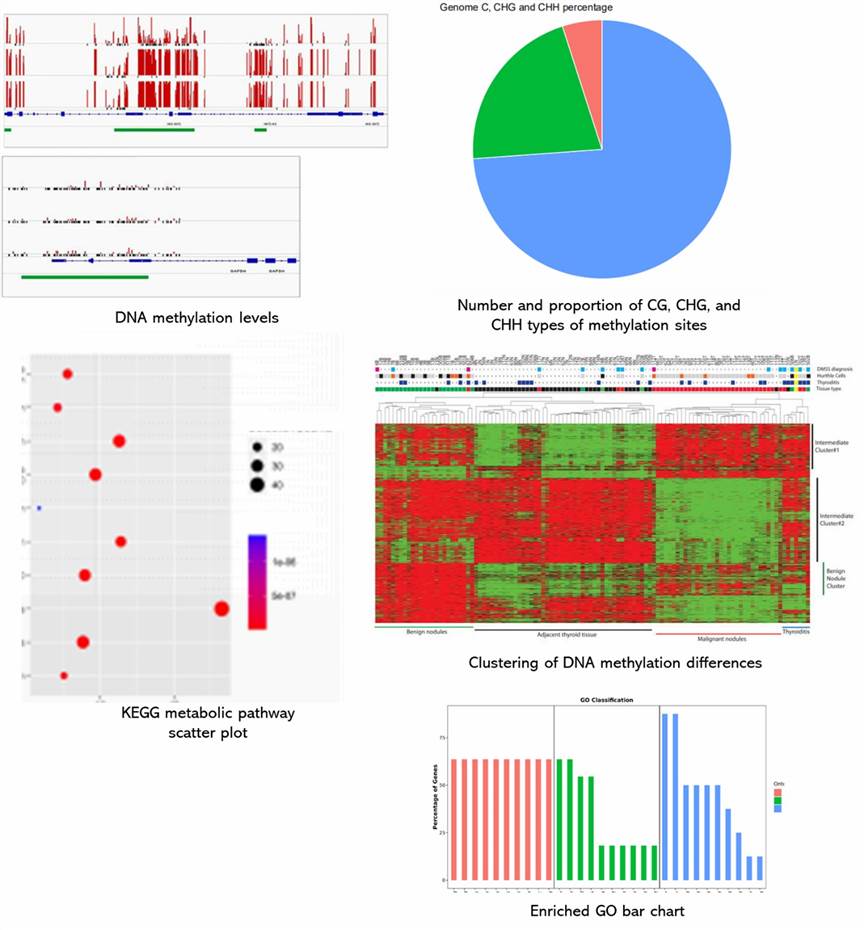
Demo Results
Partial results are shown below:
Reduced Representation Bisulfite Seq FAQs
1. What is the workflow for RRSB?
The workflow of RRBS is demonstrated in Figure 1 below. Briefly, the DNA is first digested, typically with MspI which is methylation insensitive and cuts at CCGG sites, enriching CpG rich regions of the genome. The fragment ends are then repaired and dA-tailed and adapters are ligated. Subsequently, the fragments are selected by size, converted by bisulfite, amplified and sequenced.
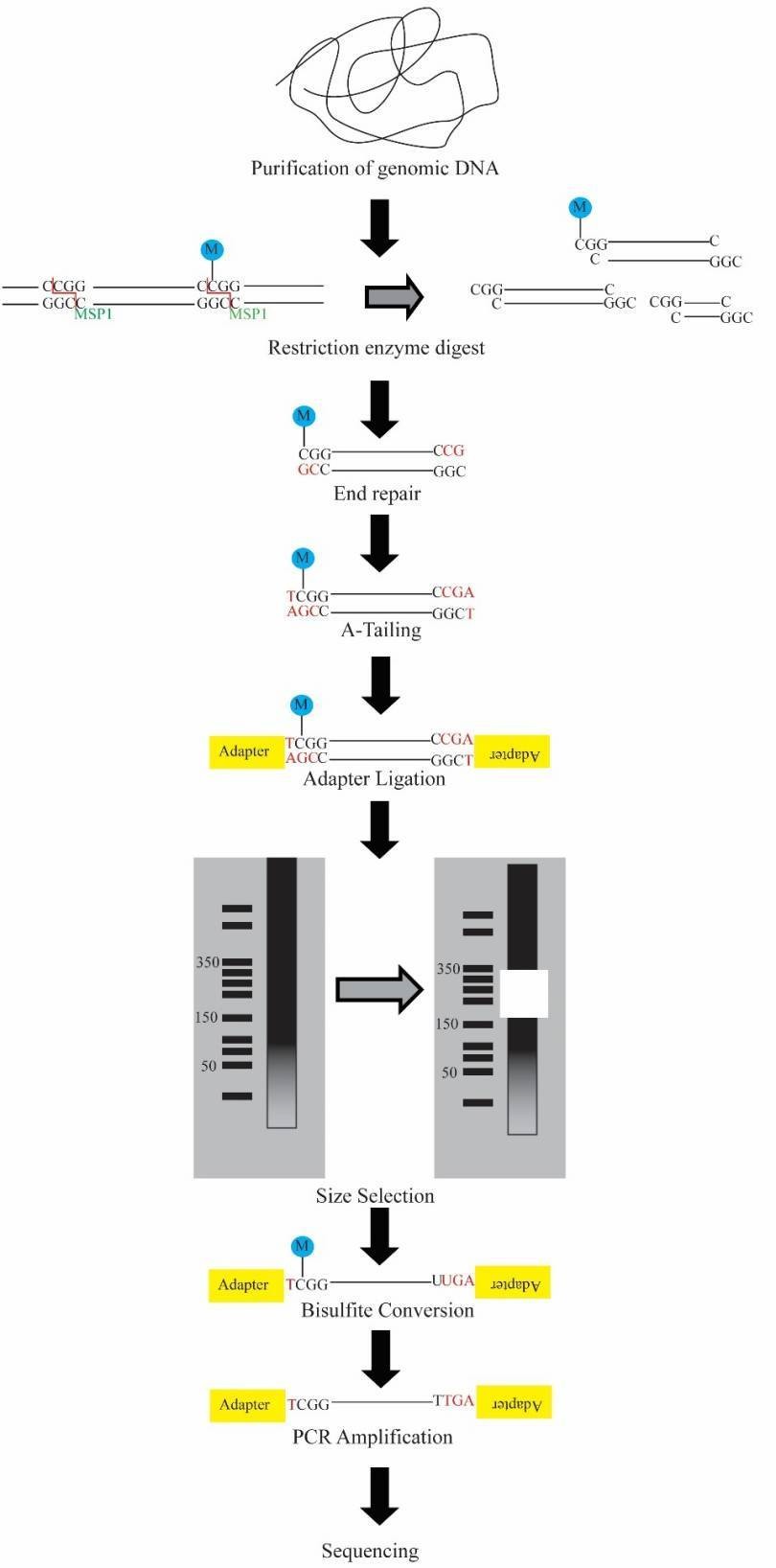 Figure 1. The schematic workflow for RRBS.
Figure 1. The schematic workflow for RRBS.
2. What are the requirements for sequencing samples?
You can submit cells (at least 5*106), fresh tissues (at least 2~3 g), or DNA (at least 5 μg; concentration ≥ 100 ng/µl; OD = 1.8~2.0; without degradation or RNA contamination or severe degradation). Before shipping, the cells should be stored at -80°C, and the DNA should be dissolved in water and stored at -20°C. Please avoid repeated freeze-thaw cycles. When shipping, the samples should be sealed with parafilm and kept in a frozen state probably with ice packs.
3. What species are suitable for reduced representation bisulfite sequencing?
The species subjected to RRBS should meet three requirements: (i) eukaryotes; (ii) its reference genome has been assembled to the scaffold level at least; (iii) relatively complete genome annotations.
Reduced Representation Bisulfite Seq Case Studies
DNA methylation changes induced by long and short photoperiods in Nasonia
Journal: Genome Research
Impact factor: 11.922
Published: December 15, 2015
Abstract
Wasp Nasonia vitripennis is an emerging model organism that exhibits a strong photoperiodic response, and can be used to study the molecular mechanisms underlying photoperiodic timing. The authors attempted to figure out how female Nasonia control the developmental trajectory of their offspring to survive the winter by taking advantage of RRBS method. It showed that knocking down DNA methyltransferase or blocking DNA methylation largely disrupts the photoperiodic diapause response of the wasps, which suggests the role of DNA methylation in insect photoperiodic timing.
Materials & Methods
- Wasp strains
- Wild-type strain, AsymC
- genomic DNA extraction
- Library construction
- RRBS sequencing
- Illumina HiSeq 2000 analyzer
- hmeDIP protocol
- Quality control
- Read filtering and alignment
- Differential methylation analysis
- Functional assays
Results
1. The Nasonia methylome
The authors profiled DNA methylation in genomic DNA extracted from female N. vitripennis kept in either long- or short-day conditions by utilizing RRBS. CpG methylation accounts for a large amount of overlap between the two samples in contrast to other sequence contexts (Figure 1B), which may suggest that non-CpG methylation largely represents experimental noise. CpGs seem to be either heavily or lightly methylated (Figure 1C). The majority of the methylated CpGs were located in exons, and the majority were located in introns, promoter regions and intergenic regions (Figure 1D, E).
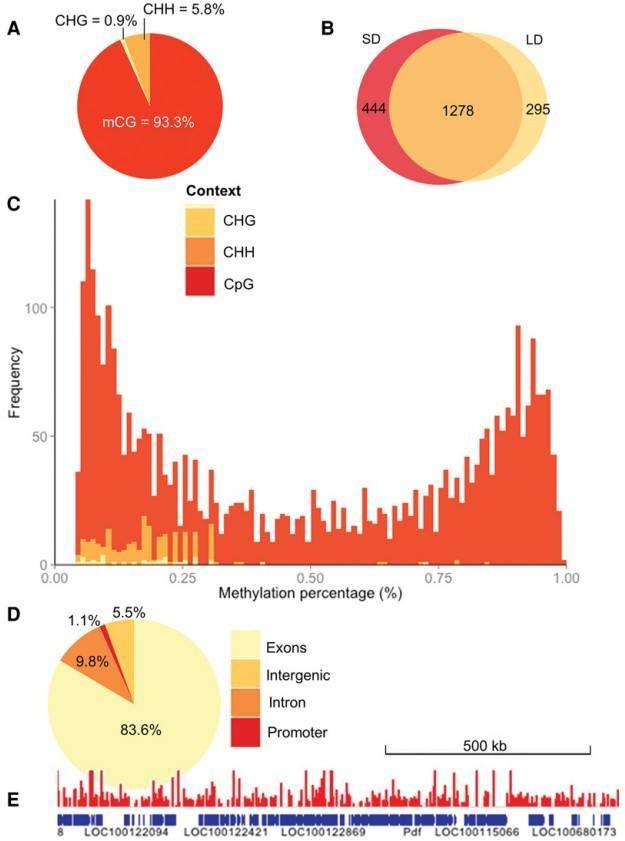 Figure 1. The Nasonia methyloma.
Figure 1. The Nasonia methyloma.
2. Identification of differentially methylated genes
The authors identified 51 differentially methylated CpG sites (DMCs) that were mapped to 37 genes. Approximately half of the DMCs (23/51) exhibited hypomethylation in long day compared with short day, whereas the others exhibited the opposite trend. qPCRs were conducted to validate the RRBS analysis.
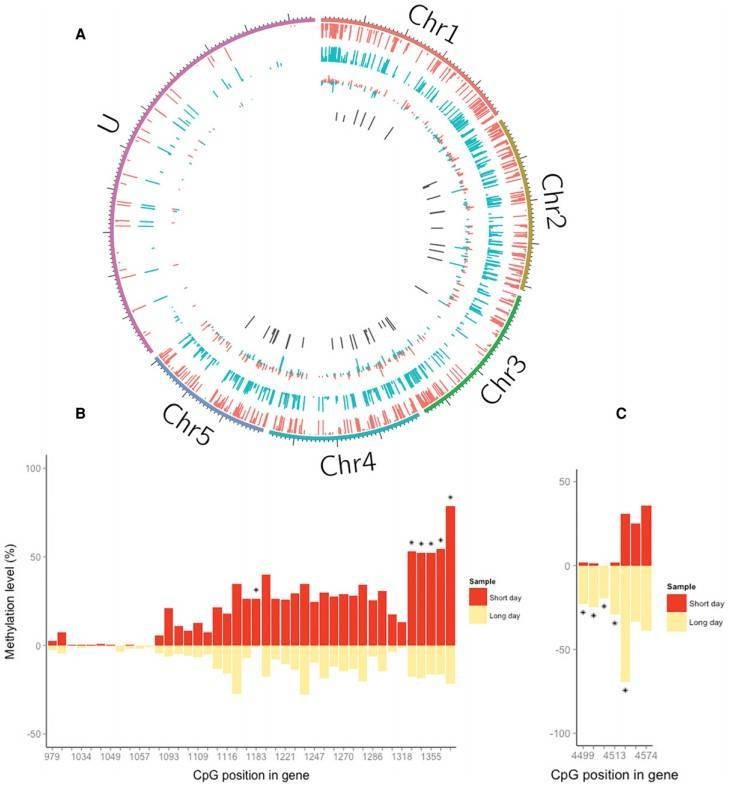 Figure 2. Differential DNA methylation associated with photoperiod in Nasonia.
Figure 2. Differential DNA methylation associated with photoperiod in Nasonia.
3. Functional assays demonstrating a causative role of DNA methylation
The authors knocked down Dnmt1a-c and Dnmt3 by injecting dsRNA into female pupae of the N. vitripennis strain AsymC. The knockdown was verified by qPCR. Knockdown of Dnmt1a led to females producing diapause offspring regardless of day length. Silencing Dnmt3 resulted a nonsignificant (but close to significance) difference in diapause response, suggesting that the effect of Dnmt3 is much weaker or harder to resolve.
They also tested the impact of DNA methylation pharmacologically by utilizing DNA methylation inhibitor 5-aza-2'-deoxy-cytidine (5-aza-dC), and observed a reduction of diapause in the progeny of wasps maintained in short day, as well as an increase in long-day progeny.
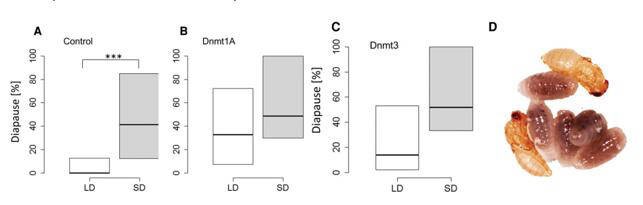 Figure 3. DNA methylation is required for photoperiodic-mediated diapause response.
Figure 3. DNA methylation is required for photoperiodic-mediated diapause response.
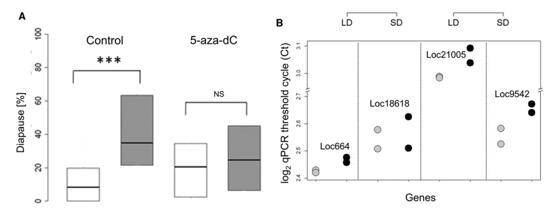 Figure 4. Pharmacological tests by using 5-aza-dC.
Figure 4. Pharmacological tests by using 5-aza-dC.
Conclusion
The authors found that DNA methylation in Nasonia vitripennis changes with the seasonal photoperiod. Disrupting key methylation enzymes (Dnmt1a or using 5-aza-dC) alters how females respond to photoperiods. Their high-resolution methylome data shows that Nasonia's methylation patterns are gene body-centric, similar to other insects. This confirms DNA methylation's role in photoperiod response and suggests that different methyltransferases have distinct functions in this process.
Reference:
-
Pegoraro M, Bafna A, Davies N J, et al. DNA methylation changes induced by long and short photoperiods in Nasonia. Genome research, 2016, 26(2): 203-210.
Related Publications
Here are some publications that have been successfully published using our services or other related services:
Folate Carrier Deficiency Drives Differential Methylation and Enhanced Cellular Potency in the Neural Plate Border
Journal: Frontiers in Cell and Developmental Biology
Year: 2022
Temporal genome-wide DNA methylation signature of post-smolt Pacific salmon challenged with Piscirickettsia salmonis
Journal: Epigenetics
Year: 2021
High-Fat Diets Fed during Pregnancy Cause Changes to Pancreatic Tissue DNA Methylation and Protein Expression in the Offspring: A Multi-Omics Approach
Journal: International Journal of Molecular Sciences
Year: 2024
Reduced representation bisulfite sequencing (RRBS) analysis reveals variation in distribution and levels of DNA methylation in white birch (Betula papyrifera) exposed to nickel
Journal: Genome
Year: 2024
Cancer-associated DNA hypermethylation of Polycomb targets requires DNMT3A dual recognition of histone H2AK119 ubiquitination and the nucleosome acidic patch
Journal: Science Advances
Year: 2024
Egr2 Deletion in Autoimmune-Prone C57BL6/lpr Mice Suppresses the Expression of Methylation-Sensitive Dlk1-Dio3 Cluster MicroRNAs
Journal: ImmunoHorizons
Year: 2023
See more articles published by our clients.


