
 Sample Submission Guidelines
Sample Submission Guidelines
2b-RAD
CD Genomics is now providing 2b-RAD sequencing which is a novel reduced-representation whole genome sequencing and restriction-site associated DNA (RAD) sequencing method for linkage mapping and determination of genome-wide variants in a cost-effective way.
What is the 2b-RAD Method
The 2b-RAD (IIB-RAD) method, initially described by Wang et al. in 2012, represents a cutting-edge approach in high-throughput SNP genotyping. Utilizing type IIB restriction endonucleases, such as Bcg I, this technique generates uniform DNA fragments ranging from 33 to 36 base pairs by cleaving at specific recognition sites. This obviates the necessity for subsequent size selection steps. One of the chief advantages of the 2b-RAD methodology is its capacity to produce consistent fragment lengths across a variety of samples, thereby facilitating precise and efficient sequencing and subsequent SNP discovery. Although the 2b-RAD method yields fewer fragments relative to single-enzyme RAD approaches, it compensates with superior resolution and coverage. Consequently, this technique addresses several inherent limitations present in earlier methods such as RAD-Seq and GBS, offering a robust alternative for comprehensive SNP analysis.
Introduction to 2b-RAD
The 2b-RAD method employs type IIB restriction enzymes (REs), such as Bcg I, which cleave genomic DNA on both sides of their recognition sites to generate fixed-size double-stranded DNA (dsDNA) fragments with protruding, non-cohesive ends. Subsequently, these DNA fragments are captured using a biotinylated adaptor specific to the initial enzyme. In comparison to analogous methods like restriction-site associated DNA (RAD) and genotyping-by-sequencing (GBS), the 2b-RAD technique offers superior specificity to regions of interest while minimizing the generation of sequences from non-informative and repetitive regions.
The consistent generation of short and uniform tags through the 2b-RAD method provides multiple advantages, including the targeting of all restriction sites, even sequencing depth across sites, high reproducibility for quantitative measurements, reduced sequencing costs per tag, and decreased sensitivity to DNA degradation. Additionally, to leverage the enhanced sequencing capacities of next-generation sequencing (NGS) platforms, we have integrated an advanced protocol that allows for the preparation of five concatenated isoRAD tags for Illumina paired-end (PE) 150 bp sequencing. This protocol grants researchers greater versatility and efficiency in designing library configurations tailored to specific research objectives.
Advantages of Our 2b-RAD Service
- Characterized by considerable flexibility, the number of tags is controllable.
- The tags exhibit consistent lengths, ensuring uniform amplification efficiency during PCR.
- These tags demonstrate positional specificity, independent of reference sequences and assembly results.
- In addition to utilizing co-dominant markers like SNPs, dominant markers can also be employed.
- Accurate and affordable
- Flexible tag number
- Consistent label length
- High density of markers
- Not requiring interim purification steps
- Allowing low DNA input and degraded DNA
- Highly reduced 2b-RAD libraries require much less sequencing for accurate genotyping
Applications of 2b-RAD
- Bin Map construction and QTL location
- Population genetic study
- Population evolution analysis
- Genome-wide association study
- Genome phylogeny
- Genomic selection
- Linkage and association mapping
- Discrimination of microbial strains
- Detection of somatic mutations
2b-RAD Workflow
The general workflow for 2b-RAD includes sample preparation, 2b-RAD library preparation, high-throughput sequencing and bioinformatics analysis. Our highly experienced expert team executes quality management, following every procedure to ensure confident and unbiased results. The library construction for 2b-RAD consists of four major stages (BsaXI digestion, ligation, amplification and barcoding).

Service Specifications
Sample Requirements
|
|
 Click |
Sequencing Strategy
|
 |
Bioinformatics Analysis
We provide multiple customized bioinformatics analyses:
|
Analysis Pipeline
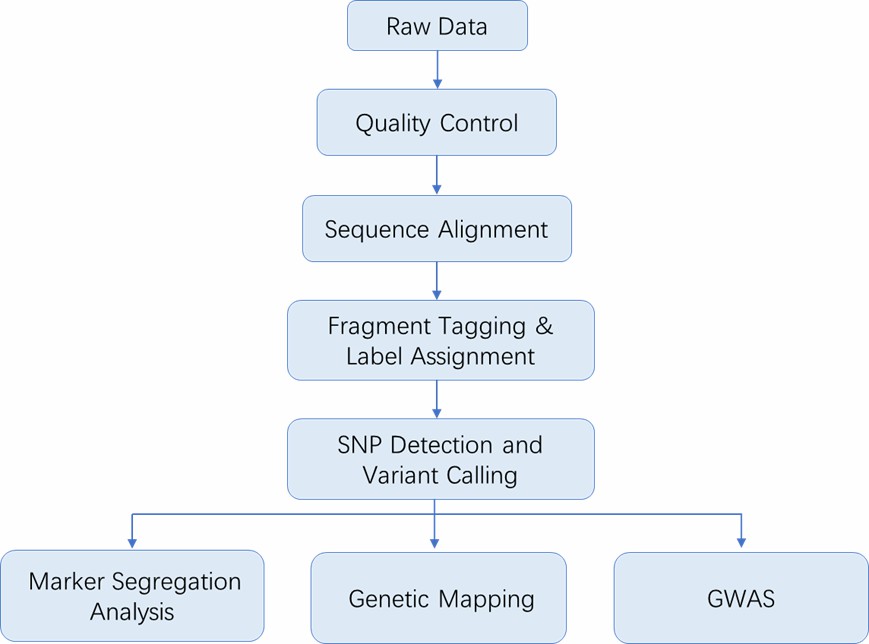
Deliverables
- The original sequencing data
- Experimental results
- Data analysis report
- Details in 2b-RAD for your writing (customization)
CD Genomics is pleased to offer cutting-edge 2b-RAD sequencing technology, providing a comprehensive service that encompasses the entire workflow from genomic DNA quality control to the determination of genotypes and the assessment of population diversity. We invite you to contact us should you have any specific needs or inquiries regarding our services. As a licensed provider of this advanced technology, CD Genomics ensures compliance and excellence in delivering robust genomic solutions. It is important to note that Keygene N.V. holds the patents and patent applications safeguarding the intellectual property associated with Sequence Based Genotyping technologies.
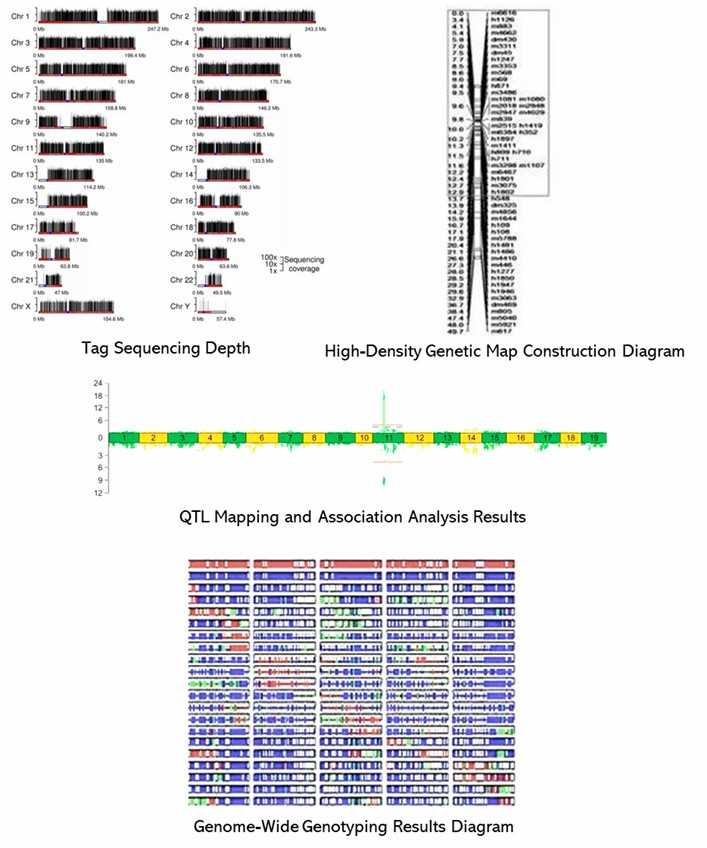
Demo Results
Partial results are shown below:
2b-RAD FAQs
1. What are the advantages of 2b-RAD?
CD Genomics is proud to offer advanced 2b-RAD sequencing technology, which harbors numerous advantages over original RAD-seq and GBS. The comparison between different reduced representation whole genome sequencing methods is outlined in Table 1.
Table 1. Technical comparison between 2b-RAD and other NGS-based reduced representation genotyping methods.
| Technology | RAD-seq, ddRAD | GBS | 2b-RAD |
|---|---|---|---|
| Library Construction | Complex, including sonication, fragment selection and multiple steps of DNA purification | No sonication and fragment selection steps, end-repair | Streamlined, no sonication and fragment selection steps, end-repair |
| Fragment Size | Non-uniform | Non-uniform | Uniform |
| Tag Density Adjustment | Difficult | Possible but not tested | Flexible |
| Tag Sequencing Depth | Highly divergent depth between different tags | Highly divergent depth between different tags | Uniform sequencing depth between tags |
| Data Analysis | False positive caused by repetitive sequences | False positive caused by repetitive sequences | Remove the distraction of repetitive sequence by using iML |
2. What are the disadvantages of 2b-RAD?
Although 2b-RAD is a powerful method for high-throughput genotyping, it also has some drawbacks. 2b-RAD works only on diploid species. Additionally, the short tags may not be long enough for locus discrimination in complex genomes.
3. How is the 2b-RAD library prepared?
The library construction for 2b-RAD consists of four major stages (BsaXI digestion, ligation, amplification and barcoding). Following DNA digestion by BsaXI, adaptors are ligated to the fragments through cohesive-end, and specific barcodes are incorporated into each sample through PCR amplification using degenerated linkers. Then the single-tag constructs are produced using modified adaptors and biotin-labeled primers, which can be further digested by SapI to generate distinct cohesive ends and then ligated in a predefined order to produce five concatenated tags. Samples are then pooled and sequenced using Illumina technology.
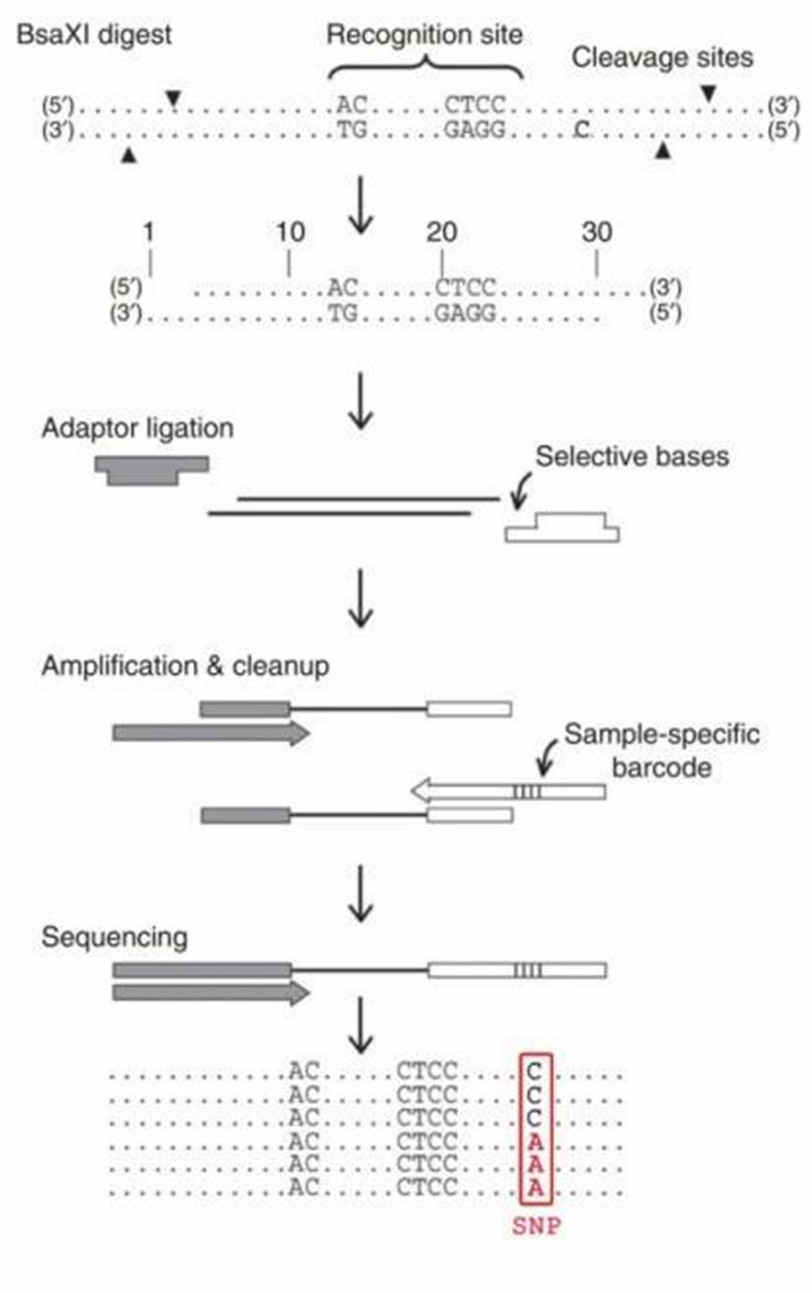 Figure 1. Schematic overview of the 2b-RAD procedure.
Figure 1. Schematic overview of the 2b-RAD procedure.
4. Can 2b-RAD detect SNP in repetitive regions?
2b-RAD may not detect SNP in repetitive regions that include a small number of restriction sites.
5. How many individuals are required for the construction of linkage map?
At least more than 100 individuals are necessary for constructing genetic linkage map.
6. For natural populations, how many samples are required?
Natural populations often have more SNP. The number of samples depends on the phenotype of interest, ranging from tens of samples to hundreds. The more samples you submit, the more accurate genotyping results you get.
7. What is the requirement for species?
Diploids with or without a reference genome are suitable for 2b-RAD. However, polyploids for 2b-RAD had better have a reference genome, and risks are still existing.
2b-RAD Case Studies
A high-resolution genetic linkage map and QTL fine mapping for growth-related traits and sex in the Yangtze River common carp (Cyprinus carpio haematopterus)
Journal: BMC Genomics
Published: 02 April 2018
Background
The authors constructed a high-resolution genetic linkage map by utilizing 7820 2b-RAD and 295 microsatellite markers in a F2 family of the Yangtze River common carp (C. c. haematopterus). They mapped a set of suggestive and significant QTLs for growth-related traits and sex on this linkage map. The genetic map and these QTL-derived candidate genes and markers are useful for further studies on the Yangtze River common carp.
Materials & Methods
Sample Preparation
- Common carp
- DNA extraction
Sequencing
- 2b-RAD sequencing
- De novo genotyping
- Microsatellite genotyping
- Linkage map construction
- Comparative genome analysis
- QTL analysis
Results
1. Construction of the high-resolution linkage map
A total of 8115 markers (7820 2b-RAD markers and 295 SSRs) were grouped into 50 LGs that was consistent to the haploid chromosome number by using the JoinMap 4.1 software.
 Figure 1. The sex-averaged genetic linkage map of the Yangtze River common carp C.c. haematopterus constructed based on 2b-RAD and microsatellite markers.
Figure 1. The sex-averaged genetic linkage map of the Yangtze River common carp C.c. haematopterus constructed based on 2b-RAD and microsatellite markers.
2. Comparative genome mapping
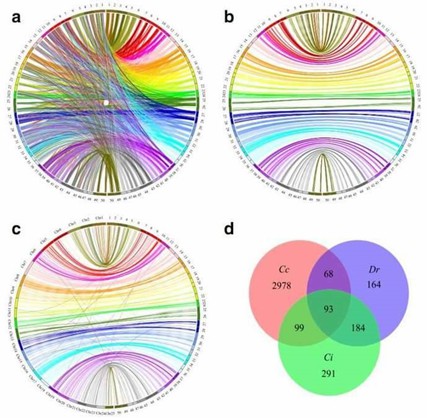 Figure 2. Circos diagram representing syntenic relationships between C. c. haematopterus (right) and (a and b) C. c. carpio (left) and Danio rerio and (d) Venn diagrams describing overlaps among uniquely aligned markers that mapped to genome of C. c. carpio (Cc), D. rerio (Dr) and C. idellus (Ci).
Figure 2. Circos diagram representing syntenic relationships between C. c. haematopterus (right) and (a and b) C. c. carpio (left) and Danio rerio and (d) Venn diagrams describing overlaps among uniquely aligned markers that mapped to genome of C. c. carpio (Cc), D. rerio (Dr) and C. idellus (Ci).
3. Fine QTL mapping for growth-related traits and sex.
A total of 21 QTLs associated with growth-related traits were detected on 12 LGs, including two genome-wide significant QTLs and 19 chromosome-wide significant QTLs.
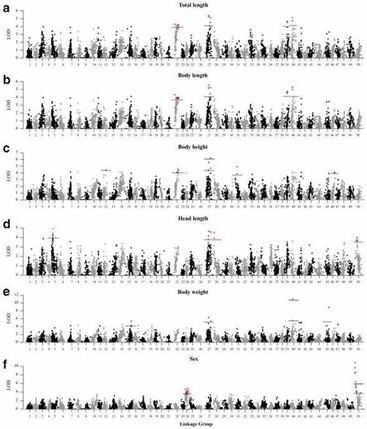 Figure 3. A genome scan of LOD profiles for (a) total length, (b) body length, (c) body height, (d) head length, (e) body weight and (f) sex in C. c. haematopterus. The dashed and solid lines indicated the chromosome-wide and genome-wide significance thresholds.
Figure 3. A genome scan of LOD profiles for (a) total length, (b) body length, (c) body height, (d) head length, (e) body weight and (f) sex in C. c. haematopterus. The dashed and solid lines indicated the chromosome-wide and genome-wide significance thresholds.
4. Potential candidate genes for growth
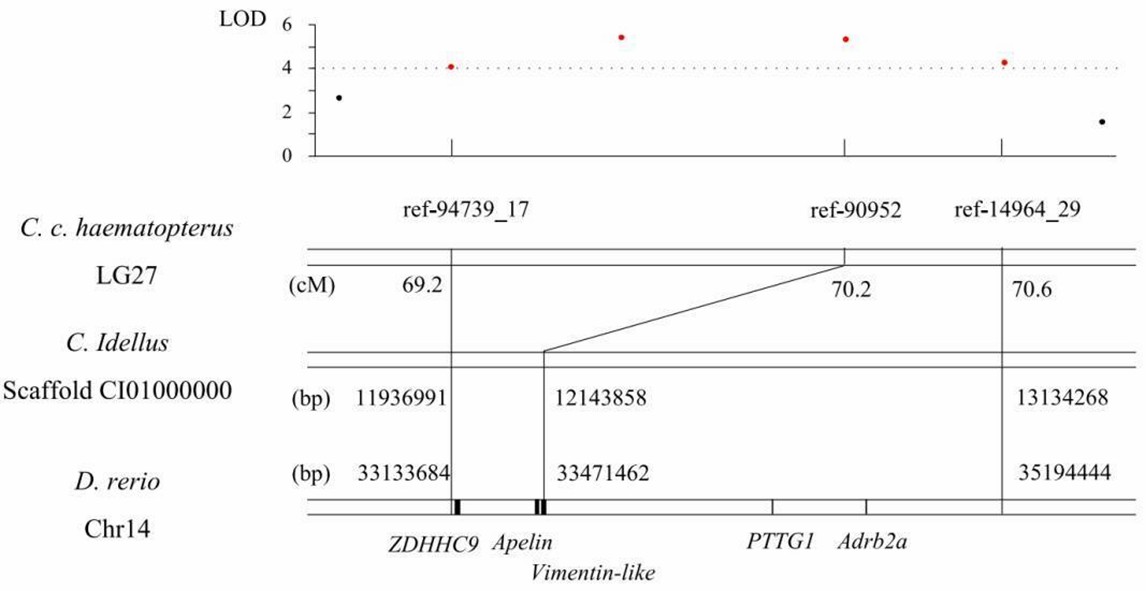 Figure 4. The QTL region for growth traits on LG27 of C. c. hasematopterus and its homologous region in genomes of D. rerio and C. idellus.
Figure 4. The QTL region for growth traits on LG27 of C. c. hasematopterus and its homologous region in genomes of D. rerio and C. idellus.
Reference
- Feng X, Yu X, Fu B, et al. A high-resolution genetic linkage map and QTL fine mapping for growth-related traits and sex in the Yangtze River common carp (Cyprinus carpio haematopterus). BMC genomics, 2018, 19(1): 230.
Related Publications
Here are some publications that have been successfully published using our services or other related services:
Drivers of genomic diversity and phenotypic development in early phases of domestication in Hermetia illucens
Journal: Insect Molecular Biology
Year: 2024
The Restriction-Modification Systems of Clostridium carboxidivorans P7
Journal: Microorganisms
Year: 2023
In the land of the blind: Exceptional subterranean speciation of cryptic troglobitic spiders of the genus Tegenaria (Araneae: Agelenidae) in Israel
Journal: Molecular Phylogenetics and Evolution
Year: 2023
Genetic Modifiers of Oral Nicotine Consumption in Chrna5 Null Mutant Mice
Journal: Front. Psychiatry
Year: 2021
A high-density genetic linkage map and QTL identification for growth traits in dusky kob (Argyrosomus japonicus)
Journal: Aquaculture
Year: 2024
Genomic and chemical evidence for local adaptation in resistance to different herbivores in Datura stramonium
Journal: Evolution
Year: 2020
See more articles published by our clients.