
 Sample Submission Guidelines
Sample Submission Guidelines
How to Study Enhancers by Sequencing?
To unravel the secrets of enhancers, scientists employ advanced sequencing techniques. Sequencing methods, such as ChIP-seq (Chromatin Immunoprecipitation with Sequencing) and Hi-C (High-throughput Chromosome Conformation Capture), enable researchers to precisely map enhancer locations, identify target genes, and understand their intricate regulatory mechanisms.
What is an Enhancer?
Enhancers are remarkable non-coding sequences residing in the genome, and they hold the key to controlling gene expression by activating target genes transcribed by RNA polymerase II (RNAPII). These essential elements are predominantly located in intergenic and intronic regions, with some even found within exons. In this article, we delve into the characteristics of enhancers and explore the methodologies used to study them through sequencing techniques.
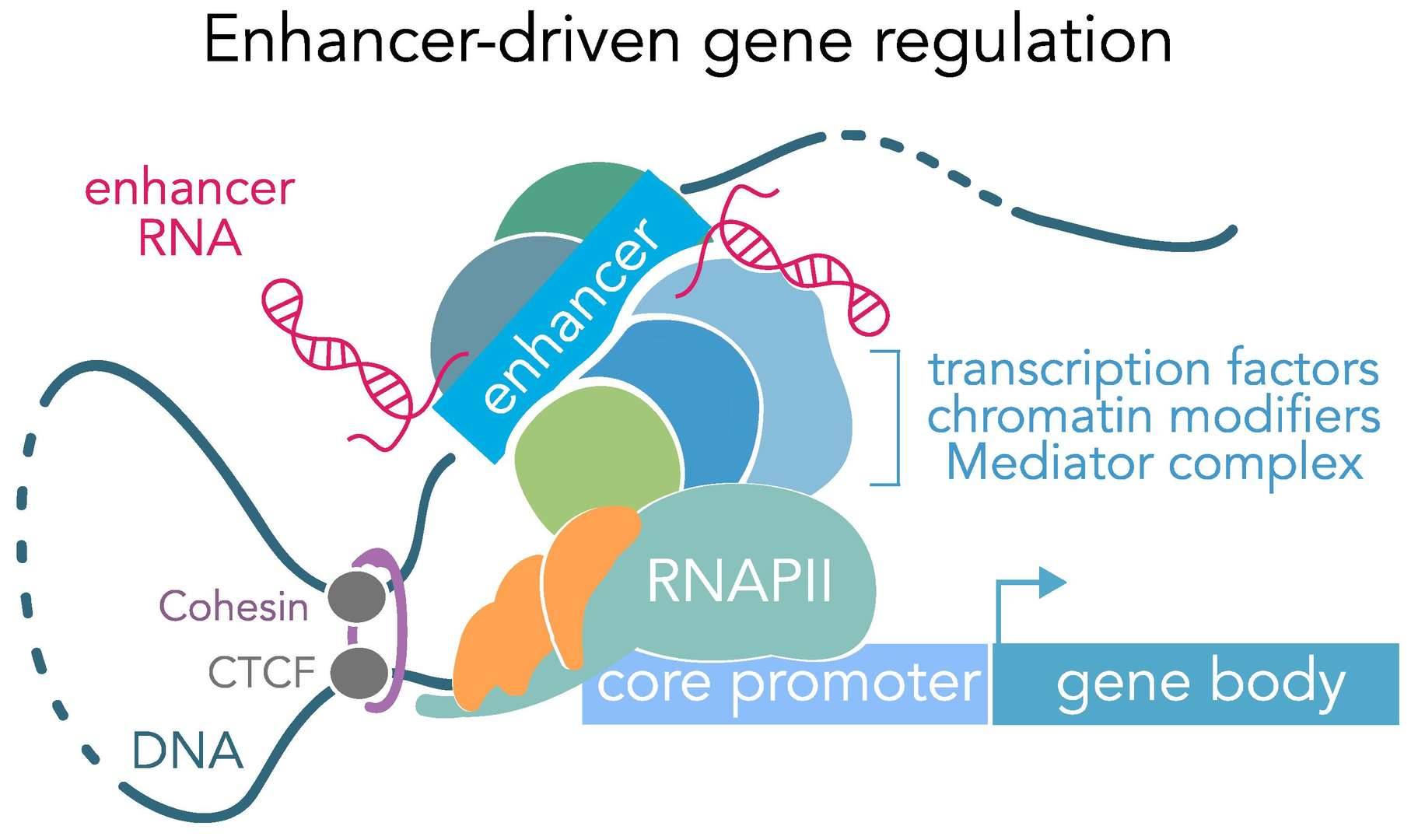 Genomic Enhancers. (Carullo et al., 2019)
Genomic Enhancers. (Carullo et al., 2019)
- Remote Regulation
One of the most fascinating aspects of enhancers is their remote nature. Typically situated upstream of the target gene at around -200 base pairs, enhancers can enhance the transcription of distant promoters. They can even activate gene expression in target genes located up to one million base pairs away. A prime example of this is the regulatory relationship between the mouse Shh gene and its enhancer ZRS sequence. ZRS, nestled within the 5th intron of the Lmbr1 gene, lies one million base pairs away from Shh. ZRS orchestrates Shh expression by forming a looping structure, a classic illustration of enhancer functionality. - Non-Directional Impact
Enhancers exhibit remarkable non-directionality in their role as transcriptional enhancers. They can effectively modulate gene expression whether located upstream, downstream, or within the target gene itself. For instance, an enhancer controlling the specific expression of the Arabidopsis gene LATERAL SUPPRESSOR (LAS) is situated 3.2 kilobases downstream of the LAS open reading frame, while the enhancer block C of the FLOWERING LOCUS T (FT) gene is positioned approximately 5 kilobases upstream of the gene. - Tissue-Specific Activity
Enhancers display distinct activity levels in various cell types or tissues. This specificity is dictated by cell- or tissue-specific protein factors. For instance, enhancers of immunoglobulin genes exhibit maximum activity exclusively in B lymphocytes. In insulin β-cells, a specific protein factor can activate the enhancer of the human insulin gene, promoting insulin gene transcription. This factor is absent in other cell types, rendering insulin gene expression primarily confined to insulin β-cells. - Phasic Properties
The functionality of enhancers is intricately connected to DNA conformation. The dynamic conformation of DNA is integral to their ability to regulate gene expression in response to various stimuli. - Heterologous Species Influence
Remarkably, some enhancers can impact gene expression across species. Mice carrying human HARE5 enhancers exhibit unique brain features found exclusively in human brains, with brain size even showing a 12% increase compared to mouse embryos with chimpanzee HARE5 enhancers. This cross-species influence highlights the versatility of enhancers in driving species-specific traits. - Responsiveness to External Signals
Certain enhancers are known to be activated by external signals. For example, steroid hormones can activate specific enhancers, and the enhancer of the heat shock gene springs into action at high temperatures, leading to the expression of the gene.
Studying Enhancers through Sequencing
- Transcription Factor Motifs
Enhancers are enriched with a diverse array of TF motifs. These motifs are specific DNA sequences that dictate the binding of TFs, triggering enhancer activation. Across various species, TF motifs have been meticulously predicted through advanced techniques like ChIP-seq (Chromatin Immunoprecipitation Sequencing) or DAP-seq (DNA Affinity Purification Sequencing). For instance, DAP-seq has successfully unveiled 529 TF motifs in Arabidopsis thaliana, bypassing the need for labor-intensive plant TF-specific antibodies and circumventing the challenges posed by low TF expression levels. Unlike ChIP-seq, which typically necessitates the construction of overexpression plasmids for tagged target proteins, DAP-seq stands out as an in vitro approach, eliminating the need for specific antibodies and making it suitable for plant species without transformation systems. - Chromatin Accessibility
The accessibility of chromatin, defined by nucleosome occupancy and chromatin-associated protein interactions, profoundly influences TF binding to regulatory sequences. Active cis-regulatory elements, such as promoters and enhancers, are predominantly found in open genomic regions known as nucleosome-deficient regions (NDRs). NDRs have been extensively mapped on a genome-wide scale in species like Arabidopsis thaliana, maize, and rice. A cutting-edge method called ATAC-seq (Assay for Transposase-Accessible Chromatin with high-throughput sequencing) has emerged as a practical alternative to conventional techniques such as MNase-seq, FAIRE-seq, and DNase-seq. ATAC-seq boasts numerous advantages, including its simplicity, reduced experimental time, and minimal sample requirements, setting it apart from its counterparts.
Please read the article ATAC-Seq – A Method to Study Open Chromatin. - Histone Modifications
Histone modifications serve as key players in gene expression regulation and chromatin accessibility. In enhancer regions, nucleosomes carry specific histone modifications that reflect their regulatory status. While animals have well-characterized histone markers, such as H3K4me1 for enhancers, H3K9ac, H4K12ac, H3K14ac, and H3K27ac for active enhancers, and H3K27me3 for inactive enhancers, plants are gradually unveiling their own intricacies. Active enhancers in plants, like those in pea (PetE) and maize (b1), are enriched in H3/H4ac and H3K9/K14ac, respectively. Additionally, intergenic NDRs in rice exhibit correlations with H4K12ac and H3K27me3. Arabidopsis research has highlighted a strong link between inactive enhancers and H3K27me3 and an even more pronounced correlation between active enhancers and H3K27ac. This implies that active enhancers in plants commonly exhibit H3 and H4 acetylation, while inactive enhancers are associated with H3K27me3. To explore these histone modifications on a genome-wide scale, ChIP-seq remains a widely-used method.
Please read the article How to Analyze ChIP-Seq Data: From Data Preprocessing to Downstream Analysis. - DNA Methylation
DNA methylation plays a pivotal role in transcriptional regulation in both animals and plants. When an enhancer bears DNA methylation, it can exert a down-regulatory influence on the expression of target genes. Notable instances include DNA methylation in the maize genes pericarp color1 (p1) and b1, and the Arabidopsis genes FLOWERING WAGENINGEN (FWA), TOO MANY MOUTHS (TMM), and FT at regulatory sequences, all of which act as suppressors of gene expression. In humans and mice, DNA methylation levels within enhancers are dynamically regulated and exhibit a negative correlation with enhancer activity. This allows for the discernment of tissue-specific enhancers. In the realm of plant biology, it has become evident that DNA methylation of cis-regulatory elements is dynamically managed, adding a layer of complexity to plant regulatory mechanisms.
Please read the article Identification of DMC, DMR, and DMG in DNA Methylation Analysis.
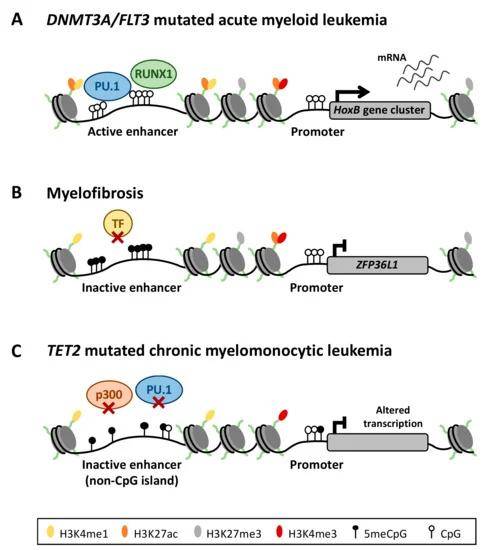 Aberrant DNA methylation of enhancer regions deregulates the transcriptional program of myeloid neoplasms. (Ordoñez et al., 2019)
Aberrant DNA methylation of enhancer regions deregulates the transcriptional program of myeloid neoplasms. (Ordoñez et al., 2019)
Low levels of DNA methylation often serve as an indicator of active enhancers. The assessment of genome-wide DNA methylation levels can be effectively accomplished through the technique of Bisulfite Sequencing (BS-seq). In BS-seq, DNA is treated with bisulfite, a process that converts cytosine residues (C) into uracil (U). However, 5-methylcytosine residues (5mC) remain resistant to this transformation. Consequently, DNA subjected to bisulfite treatment retains only the methylated cytosine. Following this principle, genomic DNA is bisulfite-converted, used to construct libraries, and subjected to high-throughput sequencing. The result is the ability to discern genome-wide methylation patterns at a single-base resolution by analyzing C-T transitions in the sequenced reads. BS-seq was originally pioneered in Arabidopsis and has since found application in several other plant species. - Chromatin Interactions
Chromatin interactions are a critical aspect of genome organization, influencing gene regulation. Techniques like Chromosome Conformation Capture (3C) and its derivatives (e.g., 4C, 5C, Hi-C, ChIA-PET, etc.) offer valuable insights into these interactions across various genomic regions. The fundamental concept behind the 3C technique involves crosslinking chromatin with formaldehyde to preserve the three-dimensional structure of chromatin. Subsequently, chromatin is cleaved using a restriction endonuclease (such as HindIII, BglII, SacI, BamH, or EcoRI). This cleavage results in the separation of interacting DNA from non-interacting genes around proteins. These interactions lead to the formation of two types of loops: those between the same genes and those involving reciprocal genes. Distinguishing between these two loop types is achieved through PCR analysis.
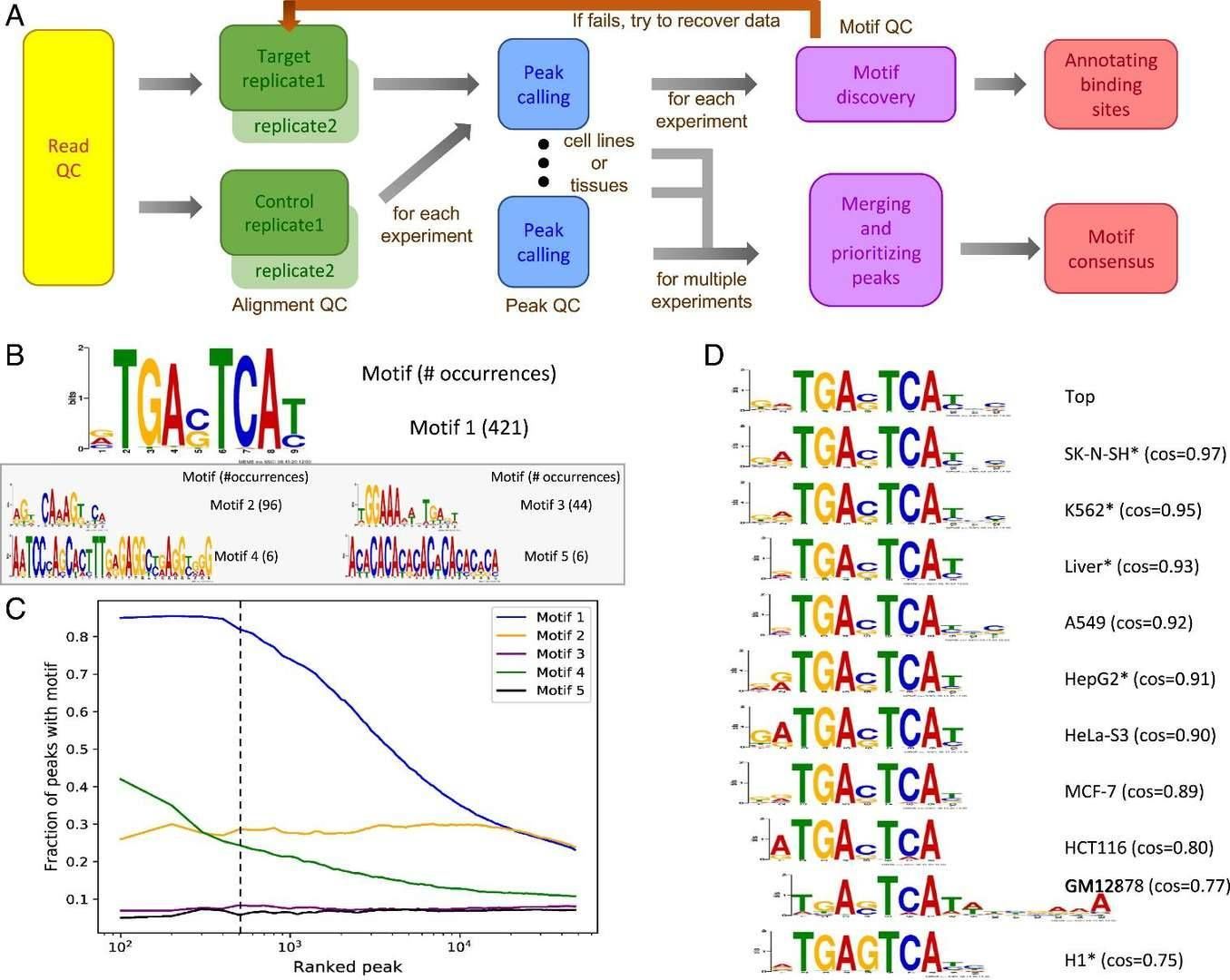 Computational pipeline and method for selecting the top PWMs of a TF from individual experiments and the top PWM across multiple experiments. (Yu et al., 2021)
Computational pipeline and method for selecting the top PWMs of a TF from individual experiments and the top PWM across multiple experiments. (Yu et al., 2021)
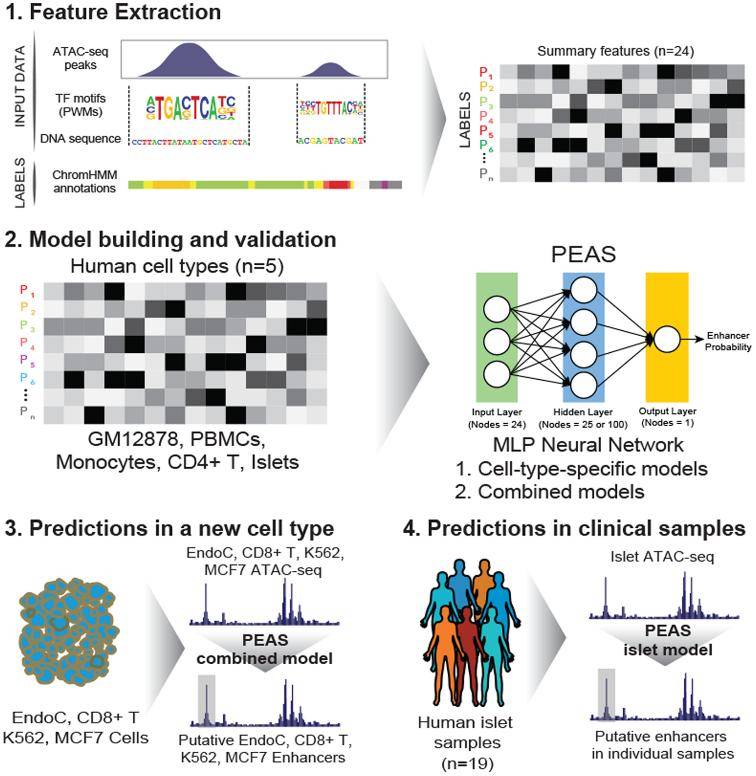 Predicts enhancers from clinical ATAC-seq samples. (Thibodeau et al., 2018)
Predicts enhancers from clinical ATAC-seq samples. (Thibodeau et al., 2018)
By employing techniques like 3C and its derivatives, researchers gain valuable insights into the spatial organization of chromatin and the dynamic interactions between genes, providing a deeper understanding of how genes are regulated in the context of their three-dimensional chromatin structure.
References:
- Carullo, Nancy VN, and Jeremy J. Day. "Genomic enhancers in brain health and disease." Genes 10.1 (2019): 43.
- Yu, Chun-Ping, et al. "Discovering unknown human and mouse transcription factor binding sites and their characteristics from ChIP-seq data." Proceedings of the National Academy of Sciences 118.20 (2021): e2026754118.
- Thibodeau, Asa, et al. "A neural network based model effectively predicts enhancers from clinical ATAC-seq samples." Scientific reports 8.1 (2018): 16048.
- Ordoñez, Raquel, et al. "DNA methylation of enhancer elements in myeloid neoplasms: think outside the promoters?." Cancers 11.10 (2019): 1424.


