
 Sample Submission Guidelines
Sample Submission Guidelines
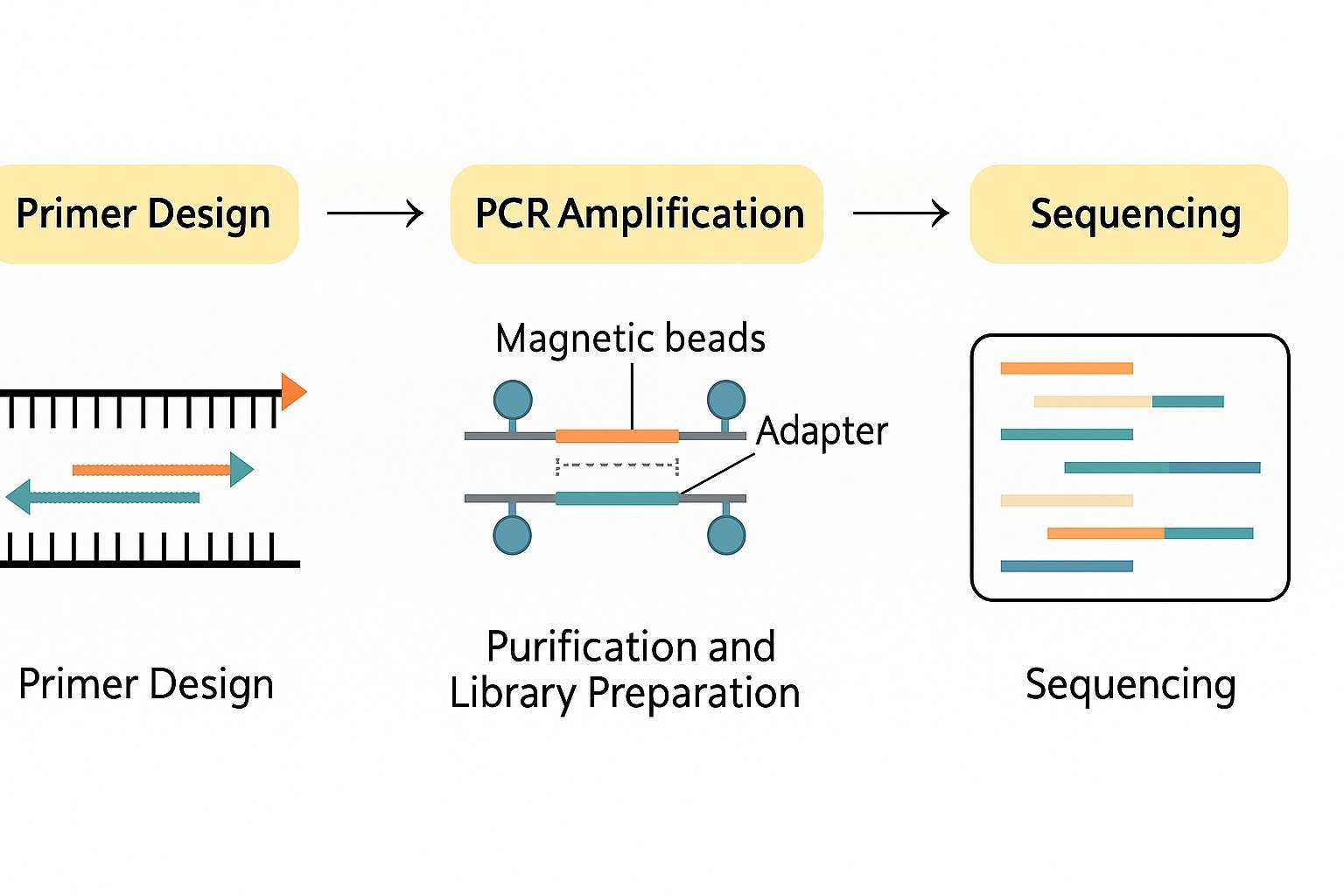
How Amplicon Sequencing Works: From Primer Design to Data Analysis
Introduction
Amplicon sequencing has transformed how researchers investigate genetic variation, microbial communities, and disease-associated mutations. Each step of the workflow - from primer design to sophisticated bioinformatics pipelines - demands meticulous optimization to ensure high data quality and biological relevance.
To learn the basic concept and significance of this technique, what is amplicon sequencing.
This article presents a detailed overview of the amplicon sequencing process, highlighting essential practices in primer design, PCR amplification, purification and library preparation, sequencing platform selection, and advanced data analysis strategies. Drawing on real-world case studies and common challenges, we provide practical insights to help researchers achieve highly efficient and accurate amplicon sequencing results across fields such as precision medicine, microbiology, and environmental surveillance.
1. Primer Design for Amplicon Sequencing
1.1 Key Parameters in Primer Design
Effective primer design is a cornerstone of successful amplicon sequencing. Several key parameters must be carefully optimized:
- Melting temperature (Tm): Tm represents the temperature at which 50% of the primer-template duplex dissociates. Ideal primer Tm values range from 55°C to 65°C, with a maximum difference of 5°C between forward and reverse primers to ensure synchronized annealing.
- GC content: A GC content between 40% and 60% balances primer stability and minimizes the risk of forming complex secondary structures. Excessively high GC content can create strong secondary structures, while low GC content may weaken binding affinity.
- Primer dimer formation and 3'-end complementarity: These must be avoided, as they can cause nonspecific amplification, consume reagents, and reduce PCR efficiency.
Primers must meet stringent requirements for specificity, structural stability, and minimal secondary structure formation. Widely used tools include Primer3, which automatically generates primers based on user-defined parameters such as length, Tm, and GC content, and BLAST, which evaluates primer specificity by identifying potential off-target hybridization.
For a detailed explanation of primer design principles and workflows, principles and workflow of 16S/18S/ITS amplicon sequencing
1.2 Software Tools and Workflow Optimization
Automated primer design tools play a pivotal role in high-throughput amplicon sequencing workflows.
- Primer3 employs sophisticated algorithms to rapidly generate optimal primers by considering multiple parameters, including primer length, Tm, and GC content.
- Geneious offers a comprehensive bioinformatics platform that integrates primer design, sequence alignment, and annotation capabilities, streamlining the entire workflow.
Laboratory protocols greatly influence how design tools and parameters are applied. Real-time experimental feedback can reveal issues such as low amplification efficiency or poor specificity. By adjusting primer design parameters accordingly - for example, modifying Primer3 settings - researchers can significantly improve primer success rates.
One laboratory case study using modular design frameworks showed that iterative optimization based on experimental feedback dramatically enhanced primer design performance.
1.3 Case Study: Bifidobacterium Community Profiling
A study published in Microorganisms (Lugli et al., 2019) reported a case where primers failed to accurately profile Bifidobacterium communities.
The primers initially designed lacked sufficient specificity, leading to unintended amplification of non-target microbial DNA, which compromised the analysis.
This case highlights the critical importance of primer specificity in microbiome research. Accurate identification and characterization of specific microbial communities depend heavily on primer performance. The 16S rRNA gene, commonly used for microbial classification, contains both conserved and variable regions. Selecting the appropriate 16S rRNA region is essential for designing primers with the necessary specificity.
You may interested in
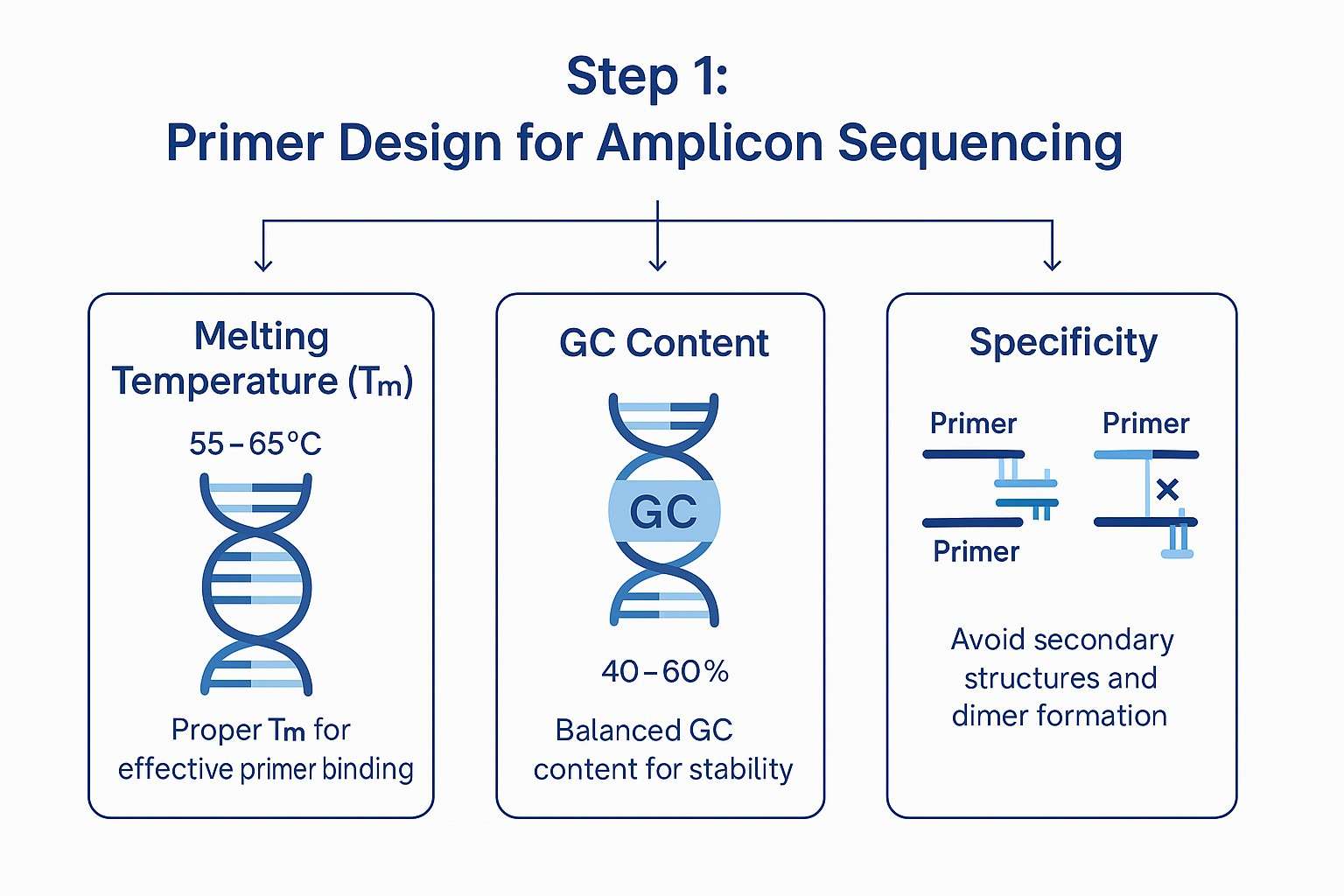 Key considerations in primer design for amplicon sequencing, including melting temperature, GC content, and specificity.
Key considerations in primer design for amplicon sequencing, including melting temperature, GC content, and specificity.
2. PCR Amplification: Creating the Amplicons
2.1 Optimizing Thermal Cycling Conditions
Thermal cycling optimization is critical to producing high-quality amplicons. A typical PCR cycle includes three key stages:
- Denaturation: Conducted at 94-98°C for 15-30 seconds, this step separates double-stranded DNA into single strands, allowing primers to bind.
- Annealing: At 50-65°C for 20-40 seconds, primers specifically bind to the single-stranded DNA template.
- Extension: Performed at 72°C, with extension times typically set at 1 minute per kilobase of target DNA.
The choice of DNA polymerase significantly impacts PCR outcomes.
- Taq polymerase is widely used due to its high thermal stability and catalytic efficiency.
- However, Taq lacks 3'-5' exonuclease proofreading activity, which may introduce errors during amplification.
For applications requiring high fidelity - such as next-generation sequencing (NGS) library construction or low-frequency mutation detection - high-fidelity polymerases like Phusion or Q5 are preferred.
Buffer composition also plays a vital role in enzyme performance, typically providing essential ions such as magnesium and potassium.
Thermo Fisher's five-step PCR workflow refines the traditional cycle by including initial denaturation, primer annealing, primer extension, amplification cycles, and final extension, allowing greater control over temperature and timing for improved efficiency and specificity.
2.2 Mitigating Amplification Bias
Amplification bias is a common issue in PCR, arising from several sources:
- GC-rich regions are difficult to denature due to strong hydrogen bonding, leading to lower amplification efficiency.
- Primer-template binding efficiency varies depending on sequence composition.
- Template secondary structures may hinder polymerase progression.
The ARTIC Network addressed these challenges in its SARS-CoV-2 amplicon sequencing protocol by employing a multiplexed primer pool design.
To better understand how amplification strategies for ribosomal RNA genes are optimized, see 16S/18S/ITS amplicon sequencing methods.
Primers were divided into multiple pools, each targeting different genome regions.
This strategy reduces primer-primer interactions, improves uniformity across amplicons, and minimizes the impact of difficult-to-amplify regions, such as those with high GC content.
Optimization of primer sequences and reaction conditions further helps balance amplification across the entire genome, resulting in more accurate sequencing data.
2.3 Quality Control for Amplicon Integrity
Strict quality control (QC) is essential to ensure amplicon purity and integrity before library preparation. Common QC methods include:
- Agarose gel electrophoresis:
This technique allows visual assessment of amplicon size and purity. Clean, sharp bands matching expected fragment sizes indicate high-quality amplification with minimal nonspecific products. - qPCR melt curve analysis:
This method monitors DNA melting behavior during gradual heating. A single, sharp melt peak suggests high specificity, while multiple peaks indicate nonspecific amplification.
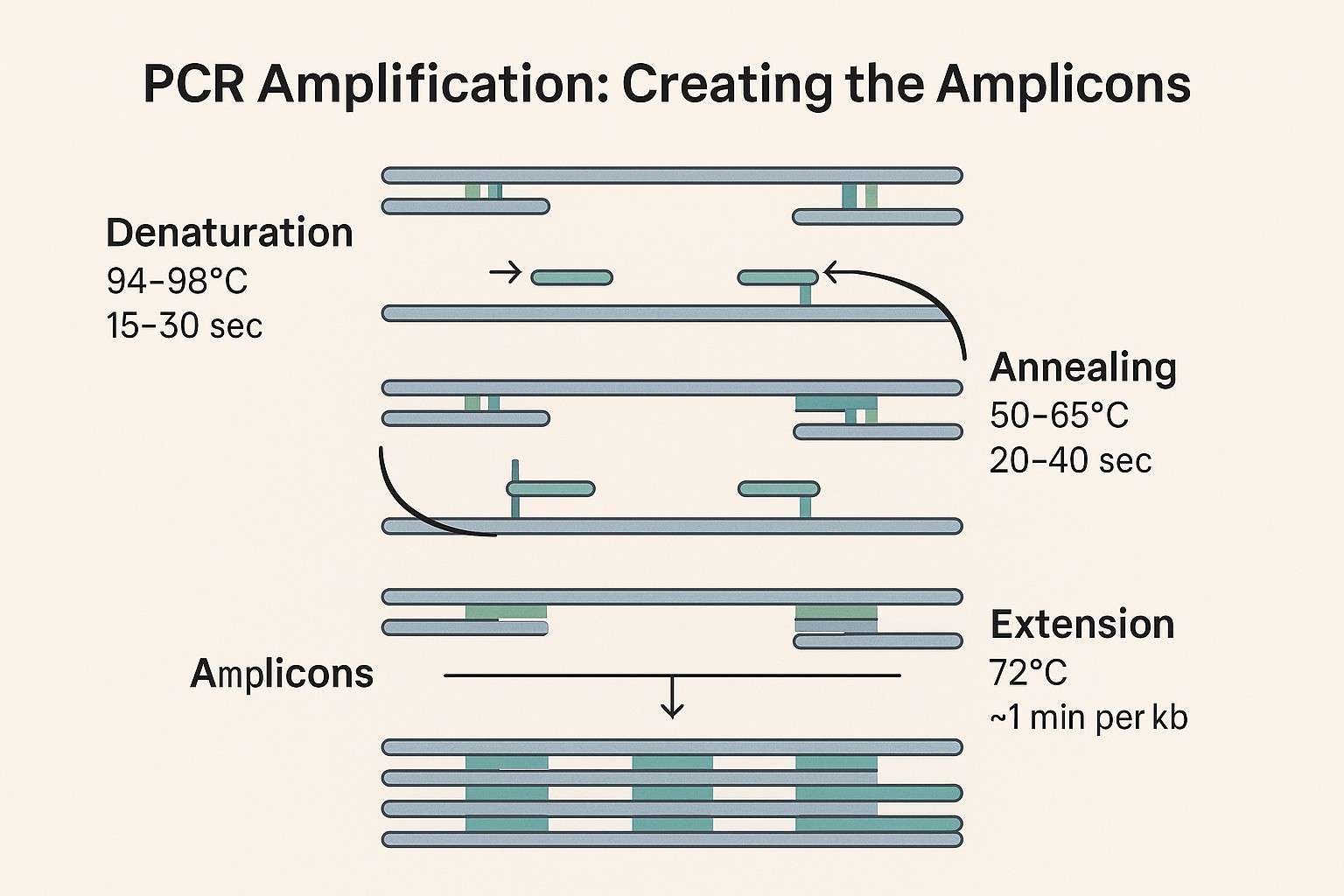 PCR amplification steps for amplicon generation, illustrating denaturation, annealing, and extension phases.
PCR amplification steps for amplicon generation, illustrating denaturation, annealing, and extension phases.
3. Amplicon Purification and Library Preparation
3.1 Magnetic Bead-Based Purification Methods
Purifying PCR amplicons is a critical step to remove contaminants and prepare high-quality DNA for sequencing. Two main methods are commonly used: magnetic bead-based purification and column-based purification, each with distinct advantages.
- Magnetic bead purification (e.g., using SPRI beads such as Agencourt AMPure XP) relies on the selective binding of nucleic acids to magnetic particles under specific buffer conditions.
Magnetic fields are then used to separate the DNA from primers, primer dimers, salts, and enzymes.
This method offers high recovery rates, excellent removal of impurities, and strong compatibility with automation, making it ideal for high-throughput workflows. - Column-based purification involves binding DNA to a silica membrane via centrifugation, followed by sequential washing and elution steps.
Although effective, this method is more labor-intensive and less amenable to automation.
It may also result in lower target recovery rates and leave trace contaminants that can interfere with downstream applications.
In comparison, magnetic bead purification provides superior target capture efficiency, scalability, and reproducibility, making it the preferred choice for large-scale amplicon purification projects.
3.2 Adapter Ligation and Indexing Strategies
Library preparation is a critical phase bridging purified amplicons to sequencing platforms.
Taking the Illumina Collibri library preparation workflow as an example, two important strategies ensure sample integrity and sequencing accuracy:
- Adapter ligation:
Specific adapters containing sequencing primer binding sites and sample-specific indices are enzymatically attached to both ends of the purified amplicons.
After ligation, libraries typically undergo PCR enrichment and size selection to amplify correctly ligated fragments and eliminate residual adapters or incomplete constructs, ensuring library consistency. - Unique Dual Indexing (UDI):
In this strategy, each sample receives a unique combination of two indices (one at each end), minimizing index-hopping artifacts and allowing precise sample identification during sequencing.
For large projects, multiple amplicon libraries can be combined (multiplexed) for sequencing.
For example, the ARTIC v3 primer pool design for SARS-CoV-2 utilized several primer pools to amplify different genomic regions. During library preparation, these distinct amplicons from different samples are proportionally pooled and sequenced together.
This approach maximizes sequencing throughput, enhances cost-efficiency, and maintains sample-specific accuracy.
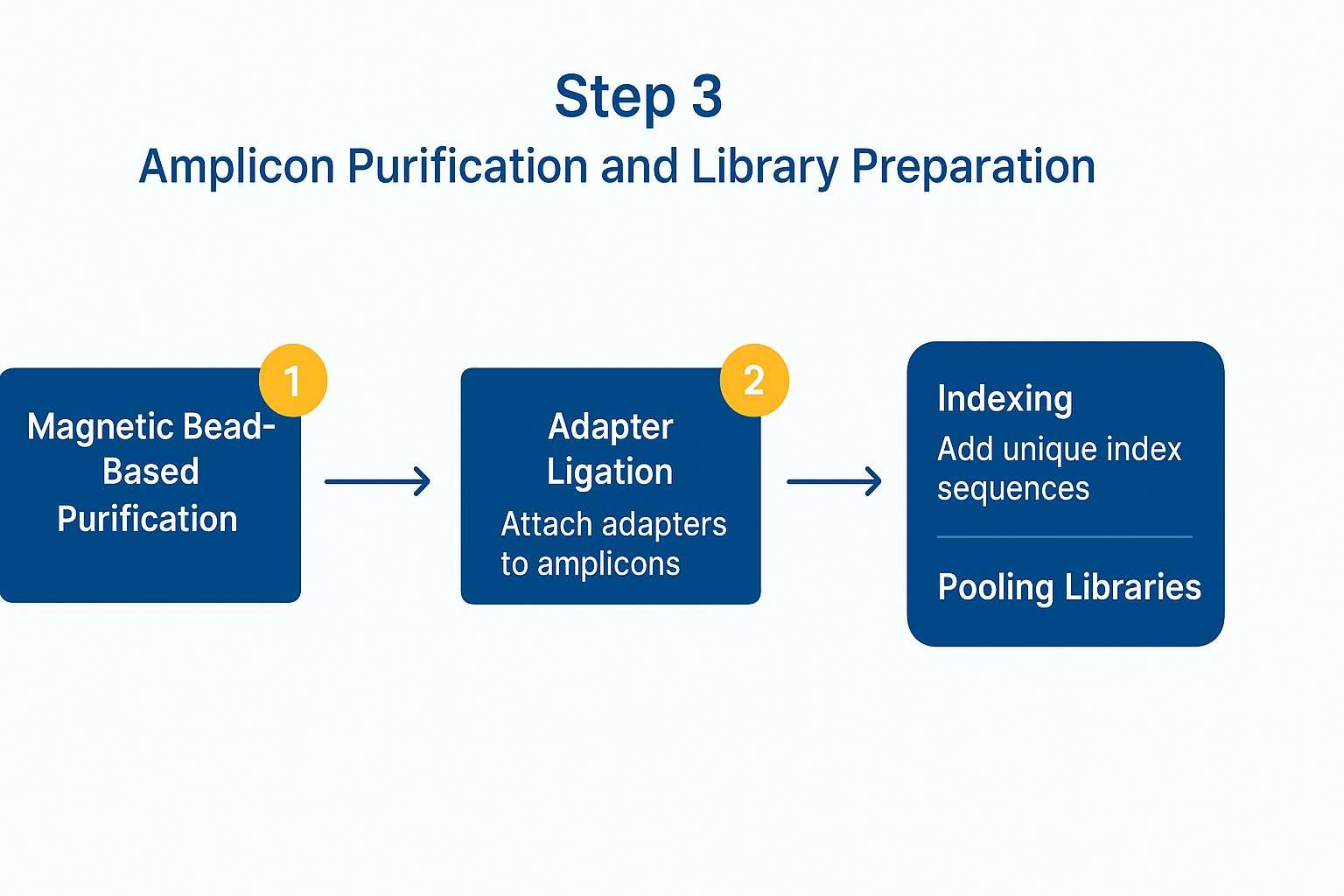 Workflow for amplicon purification using magnetic beads, followed by adapter ligation and unique dual indexing for sequencing library preparation.
Workflow for amplicon purification using magnetic beads, followed by adapter ligation and unique dual indexing for sequencing library preparation.
4. Sequencing the Amplicons
4.1 Illumina vs. Nanopore Platforms
Amplicon sequencing can be performed using a variety of next-generation sequencing (NGS) platforms, with Illumina and Nanopore technologies being two of the most commonly employed.
Each offers distinct advantages depending on project goals:
- Illumina sequencing specializes in short-read high-accuracy sequencing.
It provides exceptional coverage depth, allowing multiple redundant reads of target regions, which improves the detection of low-frequency mutations.
However, the short read length can limit its ability to resolve highly repetitive regions or complex structural variations within genomes. - Nanopore sequencing (e.g., Oxford Nanopore Technologies) enables long-read sequencing, capable of reading thousands of bases in a single pass.
This advantage allows researchers to better detect large structural variants, phase mutations across long genomic distances, and analyze complex regions more effectively.
The primary drawback of Nanopore platforms is a relatively higher error rate compared to Illumina, which can impact the accuracy of variant calling.
In practice, the choice between Illumina and Nanopore depends on project priorities:
For detecting rare point mutations, Illumina's depth and accuracy offer an advantage, while for resolving complex structural variations, Nanopore's long-read capability is indispensable.
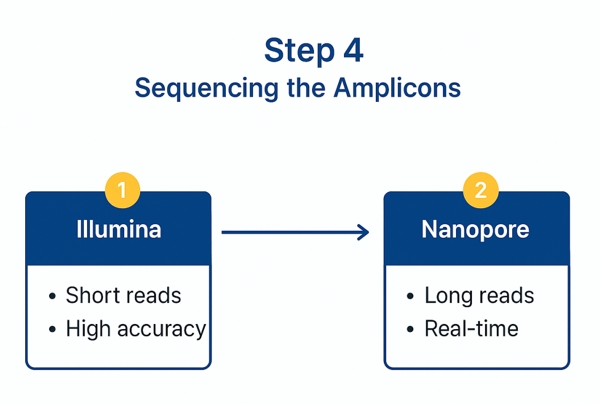 Overview of sequencing options for amplicons, comparing Illumina short-read platforms and Nanopore long-read technologies.
Overview of sequencing options for amplicons, comparing Illumina short-read platforms and Nanopore long-read technologies.
4.2 Multiplexed Amplicon Sequencing in Wastewater Surveillance
Multiplexed amplicon sequencing has demonstrated remarkable utility in public health monitoring, particularly for tracking viral pathogens in environmental samples.
A study published in Environmental Science & Technology Letters (Peccia et al., 2020) showcased this application by quantifying SARS-CoV-2 RNA in municipal wastewater systems.
Compared to traditional RT-qPCR, multiplexed amplicon sequencing provides several key advantages:
- Higher sensitivity in detecting viral genetic material, even at low concentrations.
- Simultaneous access to viral load information and comprehensive genomic sequence data.
- Early warning capabilities by detecting changes in community viral prevalence.
For example, fluctuations in viral RNA levels in wastewater samples were shown to correlate with increases or decreases in COVID-19 infection rates within local communities.
Moreover, sequencing-based approaches allowed researchers to identify emerging viral variants in sewage, enabling rapid surveillance of virus evolution and spread.
This non-invasive monitoring strategy has proven invaluable for early detection of outbreaks, providing critical data to guide public health interventions and safeguard community health.
5. Data Analysis: Interpreting Amplicon Sequencing Results
5.1 Bioinformatics Pipelines for Variant Calling
Accurate interpretation of amplicon sequencing data relies heavily on robust bioinformatics pipelines. Several tools are commonly employed for variant calling and microbial community profiling:
- QIIME2 is widely used for operational taxonomic unit (OTU) clustering based on sequence similarity thresholds.
It offers an intuitive and modular workflow but may overlook subtle sequence differences, potentially leading to less precise community resolution. - Mothur provides more sophisticated clustering by incorporating evolutionary relationships into the analysis.
This enhances taxonomic resolution compared to purely similarity-based methods.
However, both QIIME2 and Mothur have limitations when it comes to correcting sequencing errors.
To address this, DADA2 employs a model-based approach to error correction, allowing the identification of exact amplicon sequence variants (ASVs).
By eliminating noise introduced during sequencing, DADA2 significantly improves the accuracy of downstream diversity analyses and taxonomic assignments.
Integrating DADA2 into amplicon sequencing workflows has become increasingly common, particularly in studies requiring high-resolution microbial community profiling.
If you're interested in the transition from OTUs to ASVs, introduction to amplicon sequence variants.
Understanding the key differences between OTU-based and ASV-based analyses is critical for accurate interpretation of microbial diversity.
For a deeper dive into how sequencing outputs differ, refer to amplicon sequencing analysis: OTU vs ASV.
5.2 Taxonomic Classification Challenges
Despite technological advances, challenges remain in achieving accurate taxonomic classification, especially when using amplicon-based approaches.
A notable example involves the discrimination of Bifidobacterium subspecies.
The widely used 16S rRNA V4 region offers limited sequence variation among closely related Bifidobacterium strains.
As a result, subspecies-level resolution is often unattainable, leading to potential misclassification and oversimplification of microbial diversity.
This limitation arises because the V4 region is highly conserved among many bacterial taxa.
Thus, while it provides good genus- or species-level resolution for many organisms, it falls short for finer taxonomic distinctions in certain groups.
To overcome this, researchers increasingly turn to Multilocus Sequence Analysis (MLSA).
By analyzing multiple gene loci simultaneously, MLSA provides a richer dataset of genetic variation, enhancing the ability to differentiate closely related subspecies.
Applying MLSA allows for a deeper understanding of microbial community structure, ecological roles, and functional dynamics - particularly critical when studying beneficial microbes such as Bifidobacterium or when investigating complex environmental samples.
6. Conclusion: Advancements and Future Perspectives
Amplicon sequencing has made remarkable contributions to precision medicine and environmental surveillance over the past decade.
In precision medicine, it enables the sensitive detection of low-frequency mutations, facilitating early disease diagnosis and the development of personalized treatment strategies.
This targeted approach improves therapeutic efficacy and optimizes patient outcomes.
In environmental monitoring, amplicon sequencing provides a powerful tool for tracking pathogens such as SARS-CoV-2 in wastewater.
By monitoring viral load trends and detecting emerging variants, public health agencies can respond proactively to potential outbreaks, enhancing community health resilience.
Looking ahead, several emerging trends are poised to further elevate the capabilities of amplicon sequencing:
- AI-driven primer design:
Artificial intelligence algorithms are being applied to automate and optimize primer development, accounting for complex sequence contexts, minimizing off-target effects, and significantly increasing the success rate of amplification. - Single-cell amplicon sequencing:
Advancements in single-cell genomics are opening new frontiers in amplicon sequencing.
By analyzing genomic variations at the individual cell level, researchers can uncover cellular heterogeneity that would otherwise be masked in bulk analyses.
This has profound implications for understanding cancer evolution, microbial community interactions, and developmental biology.
As these innovations mature, amplicon sequencing will continue to expand its applications - driving deeper biological insights, improving clinical diagnostics, and strengthening public health surveillance systems worldwide.
For a full overview of sequencing workflows and applications, the workflow and applications of amplicon sequencing.
References:
- Lugli, G. A., Milani, C., Turroni, F., Duranti, S., Mancabelli, L., Mangifesta, M., ... & Ventura, M. (2019). Unveiling bifidobacterial biogeography across the mammalian gut using a new taxon-specific targeted sequencing approach. Microorganisms, 7(8), 328. https://doi.org/10.1038/ismej.2017.138
- Quick, J., Grubaugh, N. D., Pullan, S. T., Claro, I. M., Smith, A. D., Gangavarapu, K., ... & Loman, N. J. (2017). Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nature Protocols, 12(6), 1261-1276. https://doi.org/10.1038/nprot.2017.066
- Peccia, J., Zulli, A., Brackney, D. E., Grubaugh, N. D., Kaplan, E. H., Casanovas-Massana, A., ... & Omer, S. B. (2020). SARS-CoV-2 RNA concentrations in primary municipal sewage sludge as a leading indicator of COVID-19 outbreak dynamics. Environmental Science & Technology Letters, 7(7), 609-614. https://doi.org/10.1101/2020.05.19.20105999
- Milani, C., Lugli, G. A., Duranti, S., Turroni, F., Mancabelli, L., Ferrario, C., ... & Ventura, M. (2013). Bifidobacteria exhibit social behavior through carbohydrate resource sharing in the gut. Applied and Environmental Microbiology, 79(23), 7470-7478. https://doi.org/10.1038/srep15782
- Tyler, A. D., Mataseje, L., Urfano, C. J., Schmidt, L., Antonation, K. S., Mulvey, M. R., & Corbett, C. R. (2018). Evaluation of Oxford Nanopore's MinION sequencing device for microbial whole genome sequencing applications. Frontiers in Genetics, 9, 466. DOI: 10.1038/s41598-018-29334-5
- Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J. A., & Holmes, S. P. (2016). DADA2: High-resolution sample inference from Illumina amplicon data. Nature Methods, 13(7), 581-583. https://doi.org/10.1038/nmeth.3869


