
 Sample Submission Guidelines
Sample Submission Guidelines
A Beginner's Guide to 16S/18S/ITS Amplicon Sequencing
What is 16S/18S/ITS amplicon sequencing?
Amplicon sequencing is a targeted approach that focuses on specific regions of the DNA or RNA within a sample. It involves amplifying and sequencing a particular gene or genomic region of interest. The most commonly targeted region in microbial ecology is the 16S rRNA gene for bacteria and archaea. This gene contains conserved regions that allow for the design of universal primers, which can amplify and sequence a broad range of microbial taxa. By sequencing the 16S rRNA gene, researchers can determine the taxonomic composition of the microbial community present in the sample. Amplicon sequencing provides information about the diversity and relative abundance of different taxa but is limited in its ability to provide functional information.
If you want to know more about it, please refer to our article Principles and Workflow of 16S/18S/ITS Amplicon Sequencing.
What is the difference between metagenomics and amplicon sequencing?
Metagenomic sequencing, on the other hand, involves sequencing the entire genetic material (DNA or RNA) present in a sample, without targeting specific regions. It provides a comprehensive snapshot of the entire microbial community, including both the known and unknown microorganisms. Metagenomic sequencing can identify not only the taxonomic composition of the community but also the functional potential of the microorganisms present. By analyzing the entire genetic material, researchers can study the entire microbial genome and identify the presence of specific genes and pathways involved in various functions such as metabolism, antibiotic resistance, and virulence. Metagenomic sequencing provides a higher resolution in terms of taxonomic identification and functional potential compared to amplicon sequencing but can be more computationally and financially demanding.
Refer to Introduction to Shotgun Metagenomics, from Sampling to Data Analysis for more information.
In summary, amplicon sequencing provides information about the taxonomic composition and relative abundance of specific microbial taxa within a community, while metagenomic sequencing provides a more comprehensive view of the entire microbial community, including taxonomic information as well as functional potential. The choice between amplicon sequencing and metagenomic sequencing depends on the research objectives, available resources, and the level of detail required for the analysis.
Sanger sequencing vs. NGS-based targeted sequencing vs. long-read sequencing
Amplicon sequencing, encompassing both Sanger sequencing and next-generation sequencing (NGS) approaches, has emerged as a powerful tool for investigating microbial communities and targeted genetic analysis. Understanding the strengths and limitations of each technique is crucial for designing effective experimental strategies and interpreting results accurately.
By employing chain termination principles, Sanger sequencing provides reliable and highly accurate sequencing data, making it an ideal choice for confirming and validating results obtained from larger-scale amplicon sequencing projects. However, due to its low throughput and time-consuming nature, Sanger sequencing is less suitable for high-throughput studies involving a large number of DNA fragments.
Harnessing the power of next-generation sequencing platforms, such as Illumina sequencing, and employing strategies like amplicon sequencing or hybrid capture to selectively enrich the regions of interest. The ability to multiplex samples, wherein multiple samples are pooled and sequenced together, significantly increases throughput and reduces per-sample costs, facilitating comprehensive analysis of complex genetic variations and diverse microbial communities.
Furthermore, it is essential to consider the emergence of long-read sequencing technologies, such as PacBio sequencing and Oxford Nanopore sequencing, in the context of amplicon sequencing. Long-read sequencing offers unparalleled advantages, especially significantly longer sequencing reads in applications such as de novo genome assembly, haplotype phasing, and characterization of intricate genomic rearrangements. Nonetheless, it is worth noting that long-read sequencing technologies generally exhibit higher error rates and have limitations in terms of throughput and cost per base.
Each method possesses its own distinct strengths and considerations, necessitating careful consideration of research objectives, sample size, desired coverage, and available resources.
Bacterial 16S rRNA
There are three types of bacterial ribosomal RNA (rRNA): 5S rRNA (120 bp), 16S rRNA (about 1540 bp) and 23S rRNA (about 2900 bp). 16S rRNA is commonly found in prokaryotic cells and has a high content and copy number (accounting for more than 80% of total bacterial RNA), easy access to templates, high functional homology and moderate genetic information. The 16S rRNAs are commonly found in prokaryotic cells and have high content and copy number (80% of the total bacterial RNA).
There are nine conserved regions and nine highly variable regions in the 16S rRNA coding gene sequence. Among them, the V3-V4 region has good specificity and complete database information, which is the best choice for bacterial diversity analysis annotation.
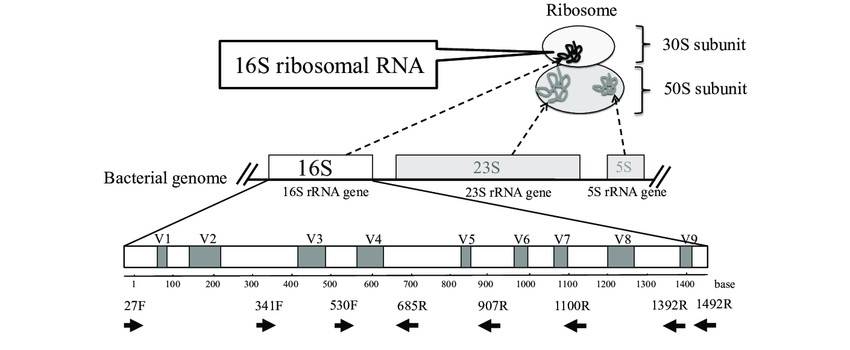 The schema of ribosome complex and 16S rRNA gene. (Fukuda et al., 2016)
The schema of ribosome complex and 16S rRNA gene. (Fukuda et al., 2016)
Eukaryotic 18S rRNA
The 18S rRNA gene is a DNA sequence encoding a small subunit of eukaryotic ribosomes, which has both conserved and variable regions (V1-V9, no V6 region). Among them, the V4 region is the most used, has the most complete database information and the best classification, and is the best choice for the annotation of 18S rRNA gene analysis.
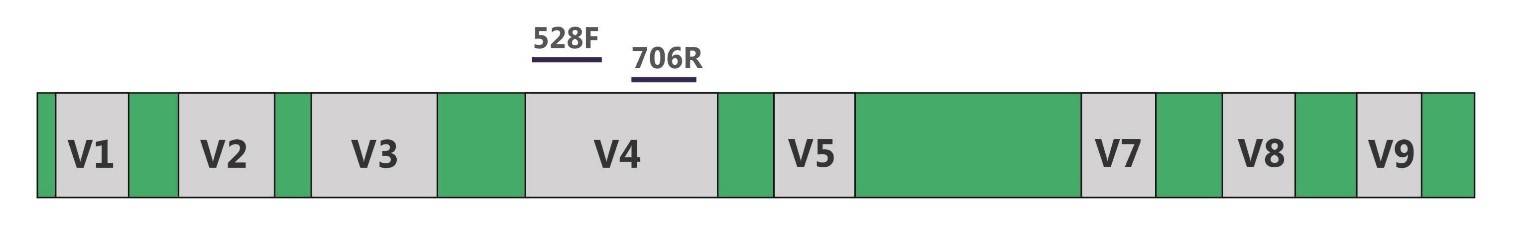 18S rRNA gene.
18S rRNA gene.
Archaeal 16S rRNA
Archaebacteria, also known as archaebacteria and archaea, are a very special class of bacteria that have some characteristics of both prokaryotes and eukaryotes. For Illumina 2×250 bp sequencing platform, primer 519F/915R is most suitable.
Fungal ITS sequences
ITS sequences are Internally Transcribed Spacer (ITS 1 and ITS 2), which are located between fungal 18S, 5.8S and 28S rRNA genes, respectively. ITS sequence fragments are small (350 bp and 400 bp for ITS 1 and ITS 2, respectively), easy to analyze, and have been widely used for phylogenetic analysis of different fungal species.
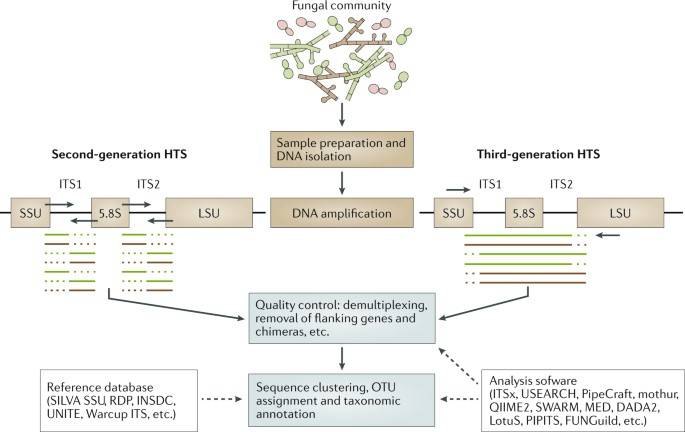 High-throughput sequencing (HTS) studies of fungal communities. (Nilsson et al., 2019)
High-throughput sequencing (HTS) studies of fungal communities. (Nilsson et al., 2019)
Diversity analysis of specific functional microorganisms
Functional microorganisms are a class of microorganisms that have received widespread attention in nature due to their functional importance, such as nitrifying bacteria, denitrifying bacteria, ammonia-oxidizing bacteria, sulfate-reducing bacteria, nitrogen-fixing bacteria, etc.
Each functional microorganism may be very different taxonomically, but has similar genes that enable them to perform the same function, so the genes that enable these functional bacteria to perform this specific function are called functional genes, such as nxrA, nirS/nirK, amoA, dsrB, nifH, nifH.
Workflow of amplicon sequencing data analysis
The downstream data are filtered to remove low-quality reads, and the remaining high-quality Clean data can be used for post-analysis; the reads are stitched into Tags by the Overlap relationship between reads; the Tags are clustered into OTUs with a given similarity, and then the OTUs are compared with the database to annotate the OTUs with species; based on the OTUs and Species annotation results were used to analyze the complexity of sample species and the species differences between groups.
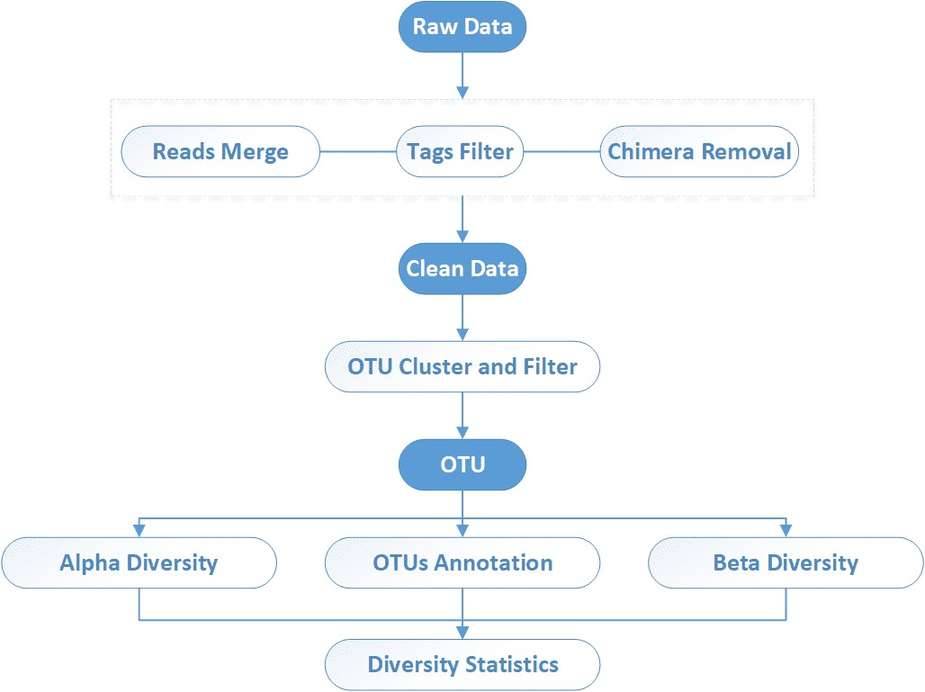 CD Genomics amplicon sequencing analysis pipeline
CD Genomics amplicon sequencing analysis pipeline
Please read our article Bioinformatics Analysis of 16S rRNA Amplicon Sequencing for more details.
Application of amplicon sequencing technology
Amplicon sequencing finds extensive applications across various fields, enabling researchers to explore and understand microbial communities in diverse environments. Some notable application areas include:
(i) Medical Field: Amplicon sequencing allows for investigating the relationship between microorganisms and human diseases. It aids in exploring the role of microbial communities in conditions such as metabolic diseases, digestive diseases, autoimmune diseases, tumor cancer, neurological diseases, and other ailments. By profiling the microbiota, researchers can identify potential biomarkers, therapeutic targets, and interventions for improved human health.
(ii) Animal Husbandry: Amplicon sequencing plays a crucial role in studying the interactions between microbial communities in the gut, rumen, and other animal-associated environments. It helps researchers explore the impact of microbiota on animal reproduction, growth and development, nutritional health, immune function, and disease treatment. By understanding the microbial ecology in animal systems, scientists can optimize animal health and productivity.
(iii) Agriculture: Amplicon sequencing facilitates the investigation of microbial interactions within the rhizosphere and other plant-associated environments. Researchers can study the effects of agricultural practices such as tillage, fertilization, and crop rotation on soil microbial communities. Understanding the interplay between microorganisms and plants aids in improving agricultural productivity, nutrient cycling, plant health, and sustainable farming practices.
(iv) Environmental Studies: Amplicon sequencing allows for the characterization of microbial communities in various environments, including polluted settings like haze, dust, residential areas, shopping malls, and natural habitats such as water bodies and marine ecosystems. It aids in understanding the distribution, composition, and dynamics of microbial populations in different ecosystems. Additionally, amplicon sequencing contributes to research on organic fertilizer fermentation and treatment, sewage treatment, oil degradation, and other environmental processes.
(v) Bioenergy: Amplicon sequencing plays a vital role in bioenergy research by enabling the identification and characterization of special functional strains and genes relevant to biofuel production. It aids in gene mining and the development of engineered bacteria with enhanced capabilities for bioenergy production.
(vi) Special Extreme Environments: Amplicon sequencing is instrumental in studying microbial taxa thriving in extreme environmental conditions, such as extreme temperatures, high salinity, acidic or alkaline pH, and other challenging settings. By analyzing the microbial diversity and functional potential in these extreme environments, researchers gain insights into the adaptation strategies of microorganisms and their role in ecosystem dynamics.
Types of samples for amplicon sequencing
Amplicon sequencing techniques, including 16S, 18S, and ITS amplicon sequencing, are applicable to a wide range of sample types.
| Sample Type | Description |
| Water (Membrane) | Freshwater, marine water, wastewater, environmental water |
| Soil | Various soil types and ecosystems |
| Intestinal | Biopsies, luminal contents, fecal samples |
| Stool | Fecal samples |
| Swab | Oral cavity, skin, nasal cavity, genital tract, etc. |
| Flora | Plant tissues, floral nectar, insect guts, ecological niches |
Please review our Sample Submission Guidelines for the most comprehensive amplicon sequencing sample collection and handling methods.
References:
- Fukuda, Kazumasa, et al. "Molecular approaches to studying microbial communities: targeting the 16S ribosomal RNA gene." Journal of UOEH 38.3 (2016): 223-232.
- Nilsson, R. Henrik, et al. "Mycobiome diversity: high-throughput sequencing and identification of fungi." Nature Reviews Microbiology 17.2 (2019): 95-109.


