
 Sample Submission Guidelines
Sample Submission Guidelines
Applications of DNA Methylation Arrays in Cancer Research
Cancer research fundamentally shapes our understanding of human well-being and mortality. The complexity of neoplastic diseases stems from diverse genomic changes combined with modifications to the epigenetic framework. Within the field of epigenetics, which governs gene regulation through biochemical modifications without DNA sequence changes, researchers have made remarkable discoveries. One key epigenetic mechanism, DNA methylation, emerges as particularly significant by regulating genetic expression through methyl group attachments.
The methylation process typically targets CpG sites, often resulting in transcriptional repression, particularly affecting genes that suppress tumor formation. Disruptions in normal methylation signatures contribute substantially to neoplastic transformation, affecting multiple cellular processes including oncogene activation and genome stability. Scientific evidence demonstrates that methylation-based changes exhibit varying effects across different cancer stages and varieties.
Contemporary analytical platforms focusing on methylation patterns have revolutionized our investigative capabilities. These advanced methodologies allow researchers to conduct extensive examinations of methylation across the entire genome, revealing previously unknown connections to cancer development. Through integration of next-generation sequencing with array-based approaches, scientists can now detect irregular methylation signatures in malignant tissues, advancing diagnostic capabilities, outcome predictions, and treatment innovations. Notable technologies include the MethyLight system, which utilizes fluorescent PCR-based detection to provide accurate quantification of methylation status in patient samples.
Understanding methylation patterns represents a cornerstone of modern cancer investigation. This growing knowledge of methylation's role in cancer biology continues driving innovations in both diagnostic tools and therapeutic interventions, ultimately advancing our ability to combat this disease effectively.
Basics of DNA Methylation Arrays
Genomic methylation analysis platforms represent cutting-edge tools for examining DNA modification patterns across the entire genome. These innovative systems employ probe-based hybridization techniques, enabling simultaneous evaluation of methylation status at numerous genomic positions. The methodology relies on strategically designed probes targeting known methylation regions, particularly CpG-rich sequences. When sample genetic material binds to these probes, researchers can measure methylation levels at specific locations. This approach facilitates extensive screening capabilities, processing vast quantities of data points within a single analytical run.
The implementation of methylation screening arrays offers distinct benefits compared to alternative approaches. These platforms have evolved into well-established, highly refined systems delivering precise and accurate methylation detection. From an economic perspective, these arrays prove particularly cost-efficient when processing substantial sample volumes. The experimental protocols remain relatively uncomplicated, minimizing the need for sophisticated preparation steps or specialized instrumentation, thus promoting widespread adoption across research facilities.
Nevertheless, certain constraints exist within this technological framework. Current array designs capture only a subset of potential methylation sites throughout the genome, providing less comprehensive coverage than next-generation sequencing methods. Although these platforms efficiently process numerous samples, extracting meaningful conclusions from the resulting data demands considerable expertise and computational resources.
These genomic screening tools maintain their position as valuable assets in methylation research, offering balanced performance between cost and capability. Despite some technical limitations, they continue serving crucial roles in understanding disease mechanisms, regulatory pathways, and clinical biomarker identification. Their practical utility makes them particularly well-suited for projects requiring efficient analysis of methylation patterns across multiple samples.
Service you may interested in
Want to know more about the details of DNA Methylation Arrays? Check out these articles:
Applications of DNA Methylation Arrays in Cancer Research
DNA methylation arrays have become indispensable in cancer research, enabling numerous applications ranging from biomarker discovery to predicting therapeutic responses. Below is an in-depth analysis of their utilization:
1. Identification of Cancer Biomarkers
How can methylation arrays aid in biomarker identification?
By comparing the methylomes of healthy individuals with those of individuals afflicted with specific diseases, differential methylation regions (DMRs) can be identified. These DMRs serve as potential biomarkers for disease diagnosis, prognosis, and prediction of therapeutic response. In cancer, DNA methylation plays a role in gene silencing; thus, aberrant methylation in promoter regions of specific genes can be used as biomarkers for early cancer detection.
Examples of Biomarkers Identified via Methylation Arrays
In bladder cancer, differential methylation of the FASLG and PRKCA genes has been utilized as prognostic biomarkers, stratifying patients into high-risk and low-risk groups based on methylation status. Additionally, in colorectal cancer, classifiers built upon hypermethylated DMRs identified through genome-wide methylation analysis have shown high accuracy in predicting recurrence. For instance, a study demonstrated that hypermethylated panels could differentiate between various adenocarcinomas with a sensitivity ranging from 77.8% to 95.9% and specificity between 91.5% and 97.7%.
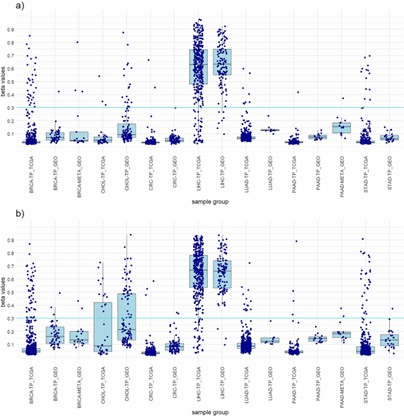 Boxplots showing the distribution of the highest beta values of all included samples from the LIHC panels of both approaches and a comparison between the TCGA dataset and the GEO dataset. (Draškovič, T., et al., 2024)
Boxplots showing the distribution of the highest beta values of all included samples from the LIHC panels of both approaches and a comparison between the TCGA dataset and the GEO dataset. (Draškovič, T., et al., 2024)
2. Understanding Cancer Development and Progression
How do methylation patterns provide insights into cancer staging?
Methylation profiles can reveal insights into various cancer stages, as tumor cells often display distinct methylation features compared to normal cells. Tumor cells typically exhibit global hypomethylation alongside hypermethylation at specific gene loci, contrasting with normal cellular methylation patterns.
The Role of Methylation in Tumorigenesis, Growth, and Metastasis
Methylation plays a crucial role in tumor initiation, growth, and metastasis. Aberrant methylation patterns can lead to the silencing of tumor suppressor genes, thereby facilitating tumorigenesis. Moreover, methylation is linked to genomic instability in tumor cells, which further drives cancer progression. Research has shown that widespread DNA methylation changes occur early in tumorigenesis and are highly pervasive across tumor types.
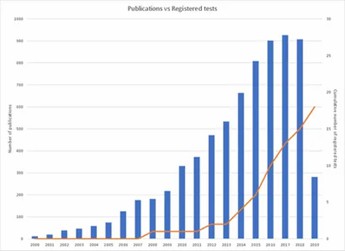 Cancer epigenetic biomarker publications per annum versus cumulative registered DNA-methylated based IVDs. (Locke, Warwick J., et al., 2019)
Cancer epigenetic biomarker publications per annum versus cumulative registered DNA-methylated based IVDs. (Locke, Warwick J., et al., 2019)
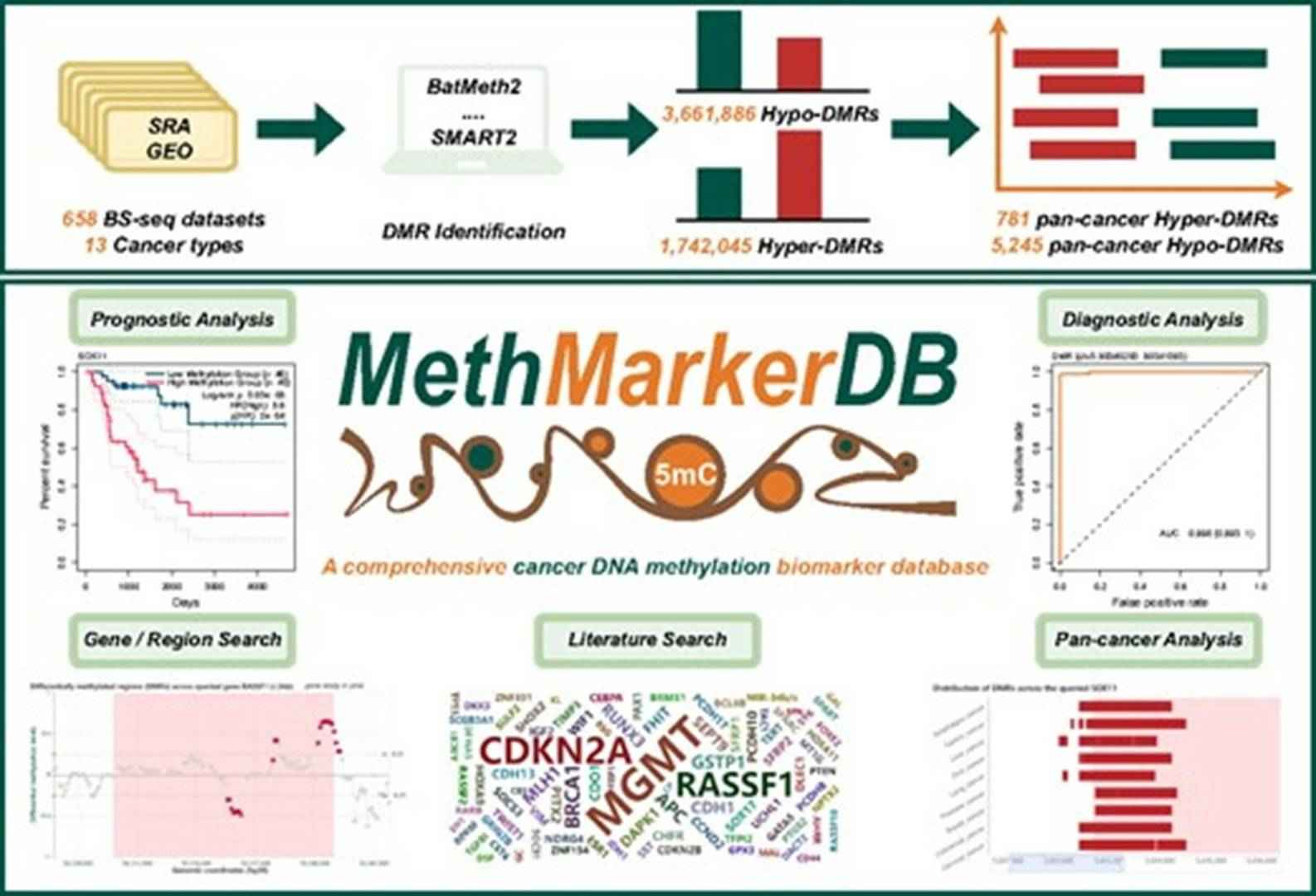 MethMarkerDB: a comprehensive cancer DNA methylation biomarker database. (Zhu, Zhixian, et al., 2024)
MethMarkerDB: a comprehensive cancer DNA methylation biomarker database. (Zhu, Zhixian, et al., 2024)
3. Classification of Cancer Subtypes
How are methylation arrays used to classify different cancer types?
Methylation arrays can differentiate cancer subtypes. For example, in breast cancer, integrating genomic DNA, RNA sequencing, and epigenetic data has enabled the identification of four primary breast cancer subtypes, with prognoses predicted based on their most unstable pathways.
Examples in Specific Cancers Such as Breast and Colon Cancer
In breast cancer, analyzing multimodal data—including mRNA, miRNA, and DNA methylation—allows for subtype differentiation. Similarly, in colon cancer, classifiers constructed from DMRs identified via genome-wide methylation analysis are used to distinguish early-stage from late-stage cancer. The integration of DNA methylation data with other genomic information has proven effective in understanding the heterogeneity within cancers.
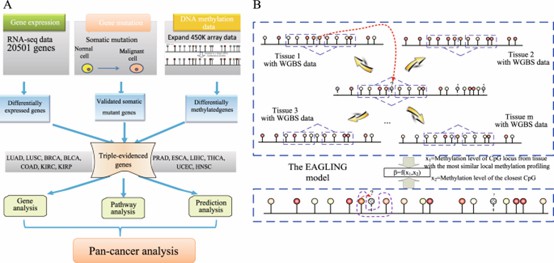 A:The multi-omics data of 13 cancers from TCGA were used to identify the genes that are differentially expressed and differentially methylated and also contain somatic mutations in each cancer. (Fan S, et al., 2019)
A:The multi-omics data of 13 cancers from TCGA were used to identify the genes that are differentially expressed and differentially methylated and also contain somatic mutations in each cancer. (Fan S, et al., 2019)
4. Prediction of Treatment Response
How can methylation profiles predict patient response to therapy?
Specific methylation patterns can alter patient responses to various treatment regimens. Changes in DNA methylation have been shown to affect drug response, making them important biomarkers for predicting therapeutic outcomes.
Studies Utilizing Methylation Arrays for This Purpose
Some studies employ methylation arrays to predict patient responses to chemotherapy. In breast cancer, alterations in DNA methylation have been used to forecast the response of triple-negative breast cancer patients to neoadjuvant chemotherapy. Furthermore, research indicates that specific hypermethylated regions correlate with treatment resistance or sensitivity across different cancer types.
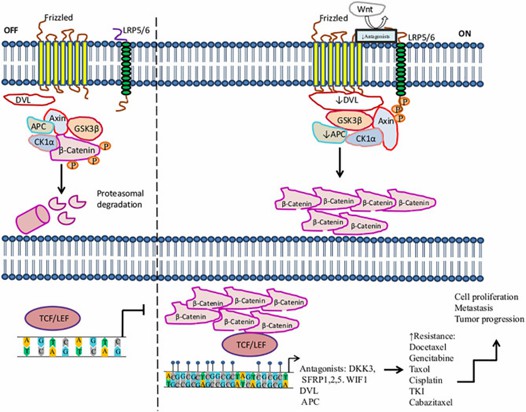 Activation of the Wnt/β-catenin signaling pathway in the resistance to therapy in cancer by methylation. (Romero-Garcia et al., 2020)
Activation of the Wnt/β-catenin signaling pathway in the resistance to therapy in cancer by methylation. (Romero-Garcia et al., 2020)
In conclusion, DNA methylation arrays offer a broad range of applications in cancer research. From early diagnosis to the prediction of treatment response, they provide critical insights and tools that further our understanding and management of cancer.
Specific Applications in Different Types of Cancer
Methylation array technology offers a significant tool in both breast and colorectal cancer research, illuminating the molecular mechanisms of cancer and fostering the development of novel diagnostic and therapeutic strategies.
1. Breast Cancer
Role of Methylation Arrays in Breast Cancer Research
Methylation array technology has become an indispensable tool in breast cancer research for identifying cancer-associated gene methylation patterns. Through these sophisticated techniques, researchers can analyze the methylation status of over 800 cancer-related genes, thereby unveiling the molecular subtypes of breast cancer. Moreover, methylation analyses are instrumental in differentiating between breast cancer subtypes, such as basal-like, Luminal A, and Luminal B, each exhibiting distinct methylation signatures.
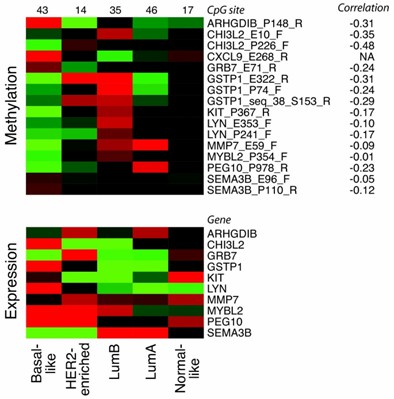 Heatmaps with average relative methylation and expression levels stratified by subtype. (Holm, K., Hegardt, C et al. 2010)
Heatmaps with average relative methylation and expression levels stratified by subtype. (Holm, K., Hegardt, C et al. 2010)
Methylation Patterns in Breast Cancer Tissues
Studies have demonstrated a strong correlation between the molecular subtypes of breast cancer and specific DNA methylation patterns. For instance, the Luminal B and basal-like subtypes exhibit higher methylation frequencies, whereas the Luminal A subtype is characterized by lower methylation frequencies. Furthermore, critical genes such as RASSF1A and BRCA1 are frequently methylated in breast cancer cell lines, suggesting that their methylation may be intricately linked with breast cancer progression.
2. Colorectal Cancer
The Application of Methylation Arrays in Colorectal Cancer Research
In colorectal cancer research, methylation array technology similarly plays a pivotal role. By scrutinizing DNA methylation patterns of colorectal cancer tissues, researchers can identify genes related to cancer progression. For example, the promoter regions of certain tumor suppressor genes often undergo methylation, resulting in gene silencing.
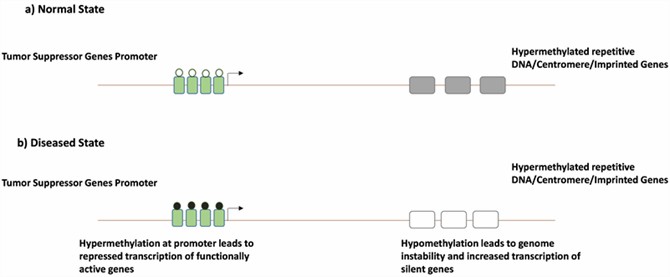 Representation of hypermethylation and hypomethylation in normal and diseased states. (Lee, Min Seob, et al., 2023)
Representation of hypermethylation and hypomethylation in normal and diseased states. (Lee, Min Seob, et al., 2023)
The Significance of Methylation Changes in Colorectal Cancer Development
Alterations in DNA methylation are fundamental to the development of colorectal cancer. Research has revealed that promoter regions of certain genes in colorectal cancer tissues commonly exhibit elevated methylation levels, whereas normal tissues display lower methylation levels. This disparity suggests that changes in DNA methylation may be a crucial mechanism in the onset and progression of colorectal cancer. Additionally, methylation analysis allows researchers to identify potential biomarkers for early cancer diagnosis and prognosis assessment.
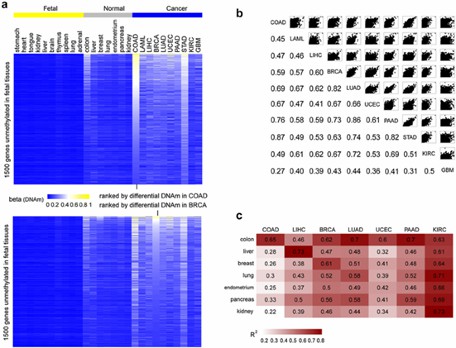 Tissue-independent cancer DNA methylation patterns. (Chen, Y et al., 2016)
Tissue-independent cancer DNA methylation patterns. (Chen, Y et al., 2016)
Challenges of DNA Methylation Arrays in Cancer Research
Technical Challenges
Researchers implementing methylation screening platforms encounter numerous technical hurdles during their investigations. Platform-specific variations and methodological differences frequently result in inconsistent measurement outcomes. While array technologies examine predetermined genomic locations, sequencing approaches provide broader coverage but face challenges regarding depth and genomic representation. Experimental variables including tissue preservation methods, nucleic acid extraction protocols, and analytical frameworks significantly impact result interpretation, making cross-study comparisons particularly challenging.
Analytical Complexities
Processing methylation datasets presents substantial difficulties for investigators. Contemporary screening methods like the Infinium platform deliver precise positional information, yet practical constraints emerge when examining diverse tumor specimens across large patient populations. Result interpretation grows increasingly complex due to methylation patterns being influenced by multiple biological factors, including cellular senescence, dietary factors, and external stimuli, necessitating careful consideration in medical applications.
Future Research Priorities
Advancing this field requires systematic efforts toward establishing standardized protocols and validation procedures, enhancing result reliability and biological significance. Investigators must develop innovative solutions addressing current technological limitations, particularly regarding large-scale sample analysis. Key priorities include improving data processing methods, minimizing technical variation, and establishing robust quality control measures. These advancements will prove essential for translating methylation research into practical clinical applications.
Future Directions
Advancing Array Technologies Through Innovation
Recent breakthroughs in screening platforms and sequencing methodologies have dramatically improved our capacity to examine genomic methylation patterns. Contemporary tools like the advanced Infinium platform featuring 935,000 distinct probes demonstrate unprecedented capabilities in oncological investigations. Supporting these hardware developments, sophisticated computational approaches continue evolving, enhancing analytical precision and processing efficiency.
Synthesizing Methylation Research with Broader Molecular Studies
Combining methylation findings with diverse molecular datasets creates opportunities for deeper biological understanding. When researchers analyze methylation patterns alongside gene expression, protein profiles, and other epigenetic modifications, novel insights emerge. This comprehensive approach proves particularly valuable in identifying cancer-specific markers and developing improved predictive frameworks for patient outcomes.
Transformative Impact on Clinical Care
Methylation analysis technologies show remarkable promise for revolutionizing medical practice. Individual methylation profiles could enable earlier disease detection, outcome prediction, and treatment optimization. These molecular signatures may guide pharmaceutical development by providing novel endpoints for therapeutic evaluation. The potential extends beyond diagnostics into treatment planning, where methylation patterns could inform strategic decisions about intervention timing and therapeutic selection.
Want to know more about the details of DNA Methylation Arrays? Check out these articles:
Conclusion
DNA methylation array technology holds promising potential in cancer research, offering a high-throughput and cost-effective means of quantitatively analyzing specific methylation sites across the genome. This capability aids in elucidating mechanisms of gene expression regulation. Despite current challenges related to technical biases and data interpretation, ongoing advancements and the integration of multi-omics data are expected to further enhance our understanding and treatment of cancer.
The potential of DNA methylation technology is substantial, not only in augmenting the precision of cancer diagnostics and therapeutics but also in providing significant biomarkers for personalized medicine. By overcoming existing technical and interpretational challenges and synergizing with other omics data, this technology is poised to bring about transformative changes in cancer research and clinical practice in the future.
References:
- Umer, Muhammad, and Zdenko Herceg. "Deciphering the epigenetic code: an overview of DNA methylation analysis methods." Antioxidants & redox signaling 18.15 (2013): 1972-1986. https://doi.org/10.1089/ars.2012.4923
- Rauluseviciute, I., Drabløs, F. & Rye, M.B. DNA methylation data by sequencing: experimental approaches and recommendations for tools and pipelines for data analysis. Clin Epigenet 11, 193 (2019). https://doi.org/10.1186/s13148-019-0795-x
- Barenboim, Maxim, et al. "DNA methylation-based classifier and gene expression signatures detect BRCAness in osteosarcoma." PLoS Computational Biology 17.11 (2021): e1009562. https://doi.org/10.1371/journal.pcbi.1009562
- Draškovič, T., Hauptman, N. Discovery of novel DNA methylation biomarker panels for the diagnosis and differentiation between common adenocarcinomas and their liver metastases. Sci Rep 14, 3095 (2024). https://doi.org/10.1038/s41598-024-53754-1
- Locke, Warwick J., et al. "DNA methylation cancer biomarkers: translation to the clinic." Frontiers in genetics 10 (2019): 1150. https://doi.org/10.3389/fgene.2019.01150
- Zhu, Zhixian, et al. "MethMarkerDB: a comprehensive cancer DNA methylation biomarker database." Nucleic Acids Research 52.D1 (2024): D1380-D1392. https://doi.org/10.1093/nar/gkad923
- Fan, S., Tang, J., Li, N. et al. Integrative analysis with expanded DNA methylation data reveals common key regulators and pathways in cancers. npj Genomic Med 4, 2 (2019). https://doi.org/10.1038/s41525-019-0077-8
- Yassi, Maryam, Aniruddha Chatterjee, and Matthew Parry. "Application of deep learning in cancer epigenetics through DNA methylation analysis." Briefings in bioinformatics 24.6 (2023): bbad411. https://doi.org/10.1093/bib/bbad411
- Romero-Garcia, Susana, Heriberto Prado-Garcia, and Angeles Carlos-Reyes. "Role of DNA methylation in the resistance to therapy in solid tumors." Frontiers in oncology 10 (2020): 1152. doi: 10.3389/fonc.2020.01152
- Lakshminarasimhan, Ranjani, and Gangning Liang. "The role of DNA methylation in cancer." DNA Methyltransferases-Role and Function (2016): 151-172. doi: 10.1007/978-3-319-43624-1_7
- Pedersen, C.A., Cao, M.D., Fleischer, T. et al. DNA methylation changes in response to neoadjuvant chemotherapy are associated with breast cancer survival. Breast Cancer Res 24, 43 (2022). https://doi.org/10.1186/s13058-022-01537-9
- Holm, K., Hegardt, C., Staaf, J. et al. Molecular subtypes of breast cancer are associated with characteristic DNA methylation patterns. Breast Cancer Res 12, R36 (2010). https://doi.org/10.1186/bcr2590
- Van der Auwera, Ilse, et al. "Array-based DNA methylation profiling for breast cancer subtype discrimination." PloS one 5.9 (2010): e12616. https://doi.org/10.1371/journal.pone.0012616
- Chen, Y., Breeze, C.E., Zhen, S. et al. Tissue-independent and tissue-specific patterns of DNA methylation alteration in cancer. Epigenetics & Chromatin 9, 10 (2016). https://doi.org/10.1186/s13072-016-0058-4


