
 Sample Submission Guidelines
Sample Submission Guidelines
The Workflow of Viral Metagenomics
How does Viral Metagenomics Work
As known to all, most of the viruses infect microorganisms, plants, and animals. They cause familiar infectious diseases (such as the flu and warts) and even some severe illnesses such as smallpox, Ebola, and HIV/AIDS. Because less than one percent of microbial hosts have been cultivated, it is very vital to identify and measure the community dynamics of viruses in the environment. Unlike bacteria and fungi, there are no evolutionarily conserved genes, so it is not feasible to monitor viral diversity using approaches analogous to 16S/18S/ITS amplicon sequencing. Viral metagenomics can provide insights into the composition and structure of viral communities.
The workflow of viral metagenomics includes sample collection, virus-like particle purification, nucleic acid extraction, library preparation, and metagenomics sequencing. It is similar to metagenomics workflow, but they differ in some steps. First, it is essential to enrich virions. Second, for RNA viruses, viral RNAs need to be converted into cDNA.
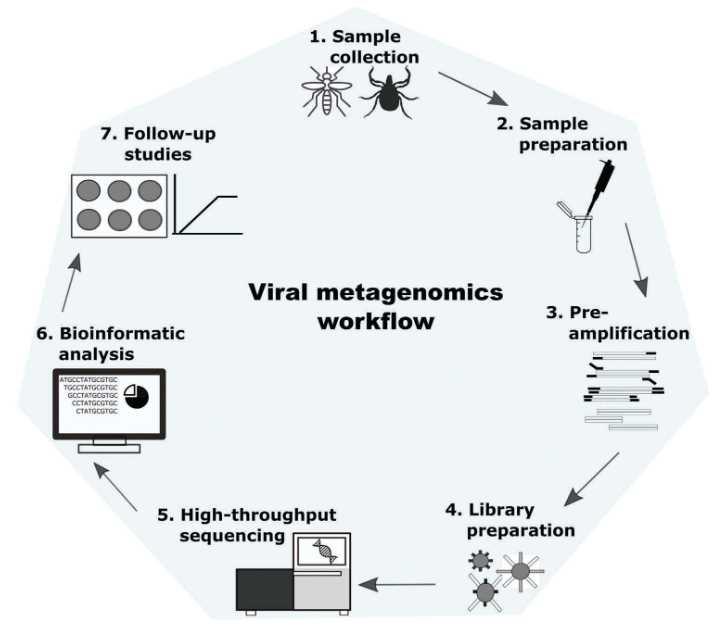 Figure 1. Workflow diagram for metagenomic analysis of viruses (Cholleti et al. 2018).
Figure 1. Workflow diagram for metagenomic analysis of viruses (Cholleti et al. 2018).
You may interested in
Viral Particle Purification Steps in Viral Metagenomics
It is complicated to isolate representative community DNA in the presence of free and cellular DNA. The average of the viral genome (~50 kb) is around 50 times smaller than the average microbial genome (~2.5 Mb). So the viral DNA signal will be overwhelmed by cellular contamination if the free DNA is not removed. Here we take sewage and clinical samples as examples to introduce how to enrich virions.
Sewage sample. Initially, 25 mL of glycine buffer (0.05 M glycine, 3% beef extract, pH 9.6) is added to 200 mL of sewage and mixed, in order to detach viral particles bound to organic material. The sample is then centrifuged at 8,000×g for 30 min. The supernatant is collected and filtered through a 0.45 μm polyethersulfone (PES) membrane to remove prokaryotic and eukaryotic cells. Viruses are precipitated from the supernatant by incubation with PEG 8000 (80 g/L) and NaCl (17.5 g/L) during agitation (100 rpm) overnight at 4˚C, followed by centrifugation for 90 min at 13,000×g. The resulting virus-containing pellet is eluted in 1 mL phosphate buffer saline (PBS) and stored at -80˚C before further processing.
Clinical sample. Before viral particle purification, it is necessary to prepare a tissue homogenate. For homogenization, a small cube of tissue (about 0.5-1 cm3) is placed in an autoclaved screw tube containing 1 mL of PBS buffer and 20-30 sterile ceramic beads. Tissue is disrupted by shaking 4 times at maximum speed at intervals of 15 s by using the FastPrep-24 Instrument. The duration of this procedure was ~0.5 h. Kohl et al. (2015) described more information on purification of viruses in clinical specimens (Figure 2).
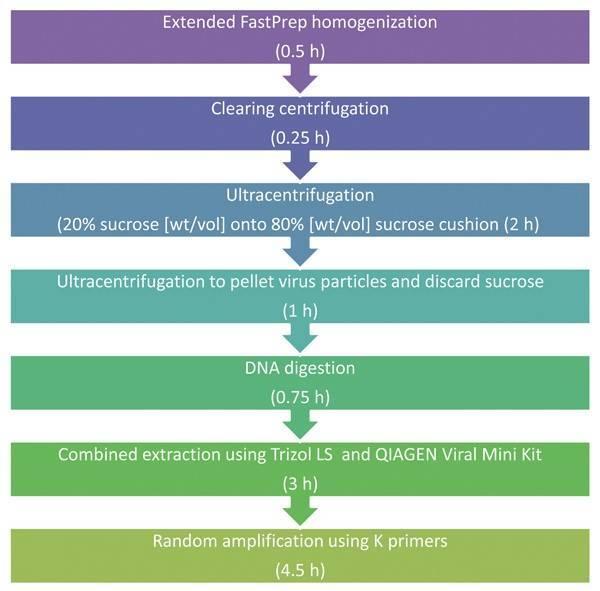 Figure 2. Schematic description of clinical based universal virus enrichment for viral metagenomics protocol (Kohl et al. 2015).
Figure 2. Schematic description of clinical based universal virus enrichment for viral metagenomics protocol (Kohl et al. 2015).
Following viral particle purification, all viral concentrates are treated with DNase I and RNase at 37°C for 15 min to remove extracellular nucleic acid, then the enzymes are deactivated at 70°C for 5 min.
Extraction of viral nucleic acid in Viral Metagenomics
In the traditional method, once the virions are isolated, the viral metagenome is then extracted and cloned. Cloning representative viral metagenomes is still challenging, due to low nucleic acid concentrations (~10-17g DNA per virion), modified DNA and the presence of lethal viral genes such as lysozymes and holins. To solve this problem, Edwards et al. (2005) developed the solution. First, it is necessary to obtain enough virions for cloning. Next, the linker-amplified shotgun library (LASL) technique makes it possible to clone small amounts of DNA and converts modified DNA into unmodified DNA via a PCR amplification step. A shearing step then disrupts lethal virus genes by fragmenting DNA into small fragments (~2 kb). It is thus possible to make representative metagenomics libraries.
Nowadays, there are many excellent kits for preparation of viral nucleic acids, such as QIA (Qiagen), NUC (Macherey-Nagel), MIN (BioMerieux), and POW (MO BIIO). For RNA viruses, the RNA needs to be converted into cDNA. Alternatively, RNA viral genome can be purified with TRIzol-LS® Reagent and DNA viral genome can be purified via phenol extraction from purified viral particles. The concentration of purified viral DNA/RNA should be quantified using RiboGreen® or PicoGreen® technologies. If the amount of nucleic acid is not sufficient for further analysis, amplification of the DNA/cDNA can be achieved by using a whole genome/transcriptome amplification kit.
Metagenomic Sequencing and Bioinformatics Analysis in Viral Metagenomics
NGS library preparation is performed using proper kits which depend on the chosen sequencing platform. The common platforms for viral metagenomics sequencing include Illumina MiSeq and HiSeq, PacBio RS II, et al. The basic bioinformatics pipeline for viral metagenomics include quality checking (identification and removal of sequence duplicates and poor-quality bases), host genome mapping (using several short reads alignment tools such as SOAP2, BWA, and Bowtie2), and taxonomic classification of viral sequences.
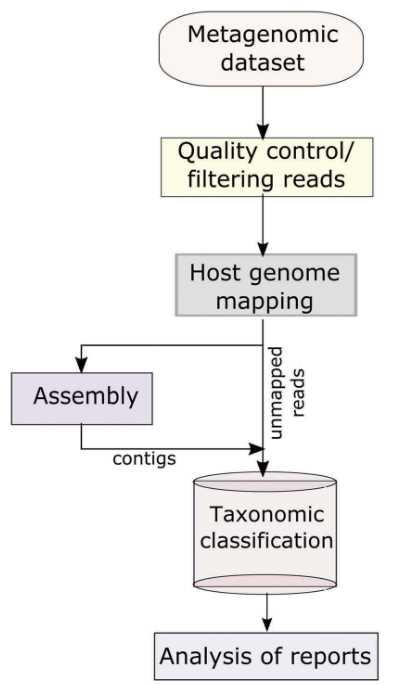 Figure 3. The basic informatics analysis of viral metagenomics (Cholleti et al. 2018).
Figure 3. The basic informatics analysis of viral metagenomics (Cholleti et al. 2018).
References:
- Cholleti H, Berg M, Hayer J, et al. Vector-borne viruses and their detection by viral metagenomics. Infection Ecology & Epidemiology, 2018, 8(1): 1553465.
- Hjelmsø M H, Hellmér M, Fernandez-Cassi X, et al. Evaluation of methods for the concentration and extraction of viruses from sewage in the context of metagenomic sequencing. PLoS One, 2017, 12(1): e0170199.
- Kohl C, Brinkmann A, Dabrowski P W, et al. Protocol for metagenomic virus detection in clinical specimens. Emerging infectious diseases, 2015, 21(1): 48.
- Parras-Moltó M, López-Bueno A. Methods for Enrichment and Sequencing of Oral Viral Assemblages: Saliva, Oral Mucosa, and Dental Plaque Viromes//The Human Virome. Humana Press, New York, NY, 2018: 143-161.