
 Sample Submission Guidelines
Sample Submission Guidelines
Analyzing Large Microbiome Datasets for Cnidarian Community Structure, Specificity, and Stability
Background
Microorganisms play an indispensable role in the survival of cnidarians, including corals. Understanding the factors influencing the microbiomes of cnidarians necessitates numerous comparative studies. However, the consolidation and analysis of diverse datasets remains a challenging endeavor.
With the growing availability of DNA sequencing data for these species and the escalating interest in coral reef health, an increasing number of studies have recognized the pivotal role of microbiomes in coral reef formation and the maintenance of ecosystem resilience. This impact encompasses essential aspects such as nutrient cycling, antimicrobial resistance, competitive exclusion of the host, growth and development, and responses to environmental changes. While the majority of these investigations have primarily focused on 16S rRNA amplification and sequencing of microbiomes in stony corals (scleractinian coral), which are significant contributors to reef-building, microbiome research within the cnidarian subclass Anthozoa, Hexacorallia, has unveiled divergent roles for other cnidarians. These alternative studies have exposed distinctive symbiotic relationships.
The lack of standardized 16S rRNA sampling, preservation, processing, and analytical protocols has imposed limitations on the amalgamation and interpretation of published microbiome data. Furthermore, the identification of distinct microbial communities in conspecific cnidarians has the potential to yield varying conclusions.
To address these challenges, bioinformatic analysis proves to be a valuable tool for effectively processing and identifying diverse and intricate microbial communities within microbiome data.
Diversity and Dynamics of Cnidarian Microbiomes
In this study, a comprehensive analysis was conducted on a substantial dataset comprising 16S rRNA sequencing data. This dataset included 12,010 cnidarian microbiomes, 3,388 sponge microbiomes, 370 seawater samples, and 245 microbiomes from artificially cultured Cordyceps spp. samples. To reduce sequencing variations, the samples were systematically categorized based on specific regions and sequencing methods.
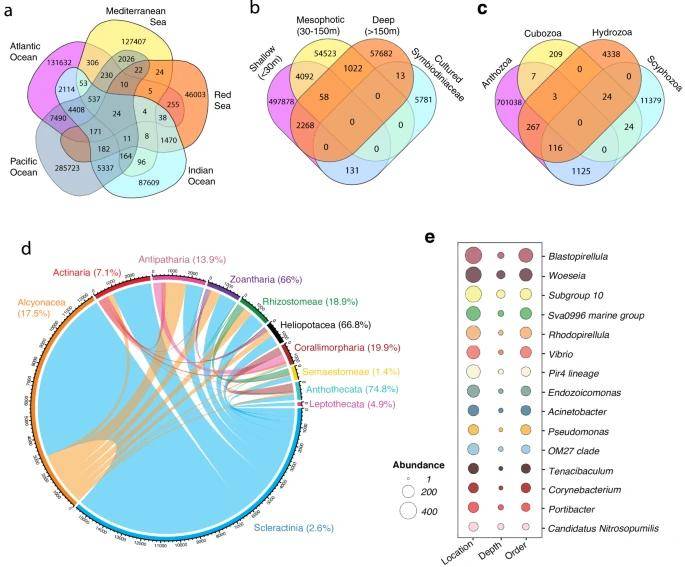 Distribution of shared microbial sequences within the phylum Cnidaria. (McCauley et al., 2023)
Distribution of shared microbial sequences within the phylum Cnidaria. (McCauley et al., 2023)
Through this rigorous integration, the analysis unveiled 86 diverse types of archaea and bacteria associated with cnidarians, identifying key species in various subphyla, depths, and microhabitats. The study highlighted the cnidarian microbiome's remarkable diversity, pinpointed significant microorganisms within it, explored key factors affecting its presence, and established a comprehensive framework for understanding the forces shaping the cnidarian microbiome.
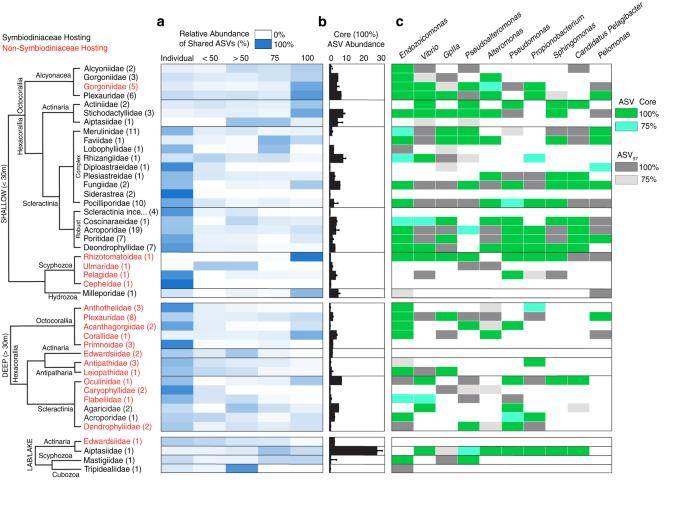 The relative abundance of archaeal and bacterial sequences identified within tissue samples per site, averaged to cnidarian family. (McCauley et al., 2023)
The relative abundance of archaeal and bacterial sequences identified within tissue samples per site, averaged to cnidarian family. (McCauley et al., 2023)
Furthermore, the investigation expanded to create a dedicated dataset for healthy cnidarian samples. This dataset was used to examine the distribution of microorganisms among these organisms. The results revealed that cnidarians in deeper waters had more distinct microbiomes, while those in shallow waters, primarily soft corals and anemones, tended to have core microbiomes with higher abundance. Additionally, the presence of symbiotic Cordyceps sinensis significantly influenced the microbial composition of cnidarians.
Comparative cluster analyses showed variations in microbiota across different microhabitats, such as tissue, bone, and mucus. Notably, the most common microbiota in tissue included Gplla, Vibrio, Pseudoalteromonas, Alteromonas, Pseudomonas, and Propionibacterium, with each found at various depths. In contrast, Sphingomonas, Pelomonas, and Candidatus Pelagibacter were more prevalent in shallow waters. Among these, Endozoicomonas emerged as a core microbial composition found in all cnidarians.
Amplicon sequence variants (ASVs) of Endozoicomonas were detected in eight cnidarian genera, including stony corals. These ASVs displayed a strong correlation with geographic distribution, with 74.3% originating from stony corals, 15.8% from soft corals, and an additional 3.8% from anemones, among which only 2.6% were shared across different taxa. Some ASVs of Endozoicomonas exhibited host-specific distribution, such as Clade 2 in Acroporidae, Clade 13 in Pocilloporidae, and Clade 14 in Rhizostomeae. On the other hand, Clades 5, 10, and 12 were found worldwide in cnidarians and sponges, spanning various microhabitats, indicating their non-host-specific nature.
Reference:
- McCauley, M., et al. "Systematic review of cnidarian microbiomes reveals insights into the structure, specificity, and fidelity of marine associations." Nature Communications 14.1 (2023): 4899.


