
 Sample Submission Guidelines
Sample Submission Guidelines
Sequencing Techniques in Identification of Bacterial Virulence Genes
Unraveling the secrets of bacterial virulence genes lies at the heart of comprehending bacterial pathogenesis, encompassing the intricacies of host infection, immune evasion, and pathogen dissemination. In this article, we delve into the forefront of scientific exploration to introduce advanced techniques and methods that empower researchers to gain a deeper and more accurate understanding of bacterial virulence genes.
Whole Genome Sequencing and Virulence Analysis
Whole genome sequencing has emerged as an indispensable tool for the discernment of bacterial virulence genes. By conducting high-throughput sequencing of a bacterium's entire genome, scientists can access intricate genetic insights, including potential virulence determinants. This reservoir of data not only aids in the prediction of drug resistance and virulence but also facilitates the identification and distribution analysis of virulence genes.
Nevertheless, standard whole genome sequencing often employs a shallow depth strategy, providing merely a cursory look into the genomes of pathogenic microorganisms present in most clinical samples. This limitation necessitates the adoption of high-depth sequencing to enhance coverage, ensuring the detection of all latent virulence genes.
A study unveiled the genetic basis for antibiotic resistance and hyper-virulence in Aeromonas veronii XhG1.2 by whole genome sequencing. And to confirm XhG1.2's identity, researchers performed 16S rDNA sequence analysis and whole genome sequencing, aligning it within the Aeromonas veronii species. Using the Virulence Factor Database (VFDB), they identified virulence genes, including aerolysin, RtxA, T2SS, T3SS, and T6SS. These factors signify XhG1.2's capacity to cause severe infections. These factors are known to play pivotal roles in the pathogenicity of various bacterial species. The presence of these virulence genes underscored XhG1.2's potential to cause severe infections in host organisms.
Equally concerning was the prediction of antibiotic resistance genes in XhG1.2, as revealed by the Comprehensive Antibiotic Resistance Database (CARD) analysis. Among the identified resistance genes were CephA3, OXA-12, adeF, and pulvomycin resistance genes. These genes signify XhG1.2's capacity to withstand various antibiotics, potentially making it a formidable adversary in the context of antimicrobial therapy.
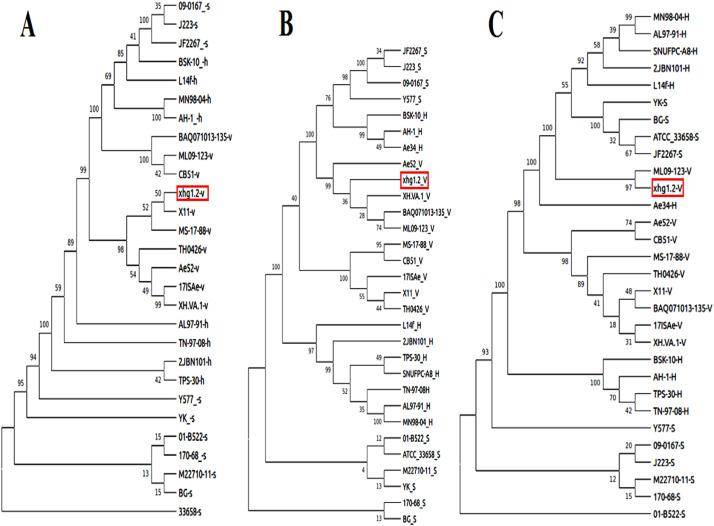 Phylogenetic-analysis of virulence factors of XhG1.2 along with sequences from databases. (Das et al., 2021)
Phylogenetic-analysis of virulence factors of XhG1.2 along with sequences from databases. (Das et al., 2021)
Metagenomic Sequencing and Improved Coverage
In addition to whole genome sequencing, metagenomic sequencing plays a vital role in deciphering the complex genetic landscapes within microbial communities. However, metagenome sequencing usually employs a shallow depth sequencing strategy, resulting in only about 0.5-fold coverage of pathogenic microorganisms in most clinical samples, thus limiting the accurate detection of virulence genes.
To overcome this challenge, researchers may consider embracing high-depth metagenomic sequencing. By implementing this approach, they can significantly improve coverage, ensuring that even the most elusive virulence genes are not overlooked. High-depth metagenomic sequencing provides a more comprehensive view of the genetic makeup of complex microbial populations, allowing for the precise identification of virulence factors and their potential contributions to pathogenicity.
Applications of Comparative Genomics
Comparative genomics stands as a potent approach, permitting the identification of virulence factors by scrutinizing DNA sequences across different bacterial strains at a genome-wide scale. This method empowers us to pinpoint shared genes associated with pathogenicity while effectively sieving out genes linked to non-pathogenicity.
Moreover, comparative genomics can unveil alterations in promoter sequences that trigger shifts in gene expression patterns, ultimately bolstering bacterial virulence. Investigating these sequence modifications enables scientists to gain profound insights into bacterial adaptations across diverse host environments.
In the case mentioned before, phylogenetic and comparative genomic analyses further solidified their findings. They discovered that A. veronii species, to which XhG1.2 belongs, shared common genes associated with toxin production. This not only reaffirmed the pathogenic nature of A. veronii XhG1.2 but also highlighted the broader implications of this strain's potential to cause disease in a diverse range of ornamental fishes.
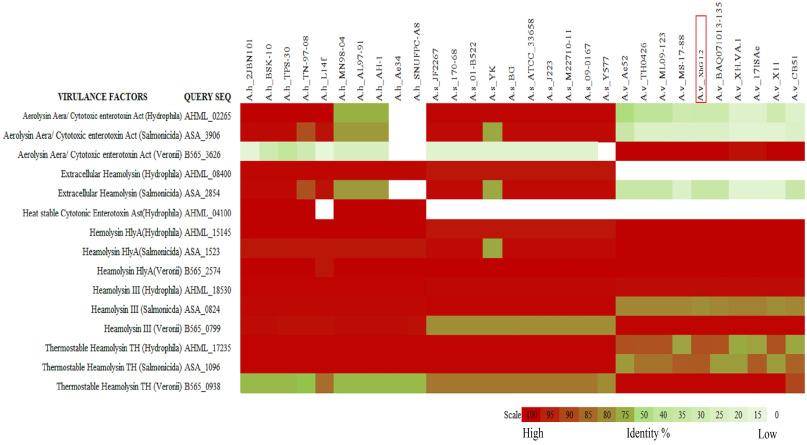 Heat map representation of comparative genome data of Aeromonas veronii against A. hydrophila, and A. salmonicida. A. veronii XhG 1.2 genome is marked under the red box. (Das et al., 2021)
Heat map representation of comparative genome data of Aeromonas veronii against A. hydrophila, and A. salmonicida. A. veronii XhG 1.2 genome is marked under the red box. (Das et al., 2021)
Virulence Assays
Virulence assays represent a pivotal step in the quest to uncover bacterial virulence genes. These methodologies enable us to evaluate bacterial behavior within the milieu of infected host cells. Typically, cellular and animal models are employed to simulate the infection process and assess the impact of virulence genes.
Within cellular models, metrics such as the bacterium's ability to infect host cells, intracellular replication rates, and cytotoxicity can be precisely quantified. This wealth of data enables the determination of the virulence effects of specific genes. Meanwhile, animal models facilitate the study of various biological aspects of bacterial infections, including disease progression, clinical signs, and pathogenicity.
Integration of Different Methods
It is crucial to underscore that bacterial virulence is an intricate interplay of multiple genes, rather than the result of a single gene's action. Thus, the harmonious integration of whole genome sequencing, comparative genomics, and virulence assays emerges as the most effective strategy for identifying bacterial virulence genes. This holistic approach offers a comprehensive understanding of the nature and functions of virulence factors, laying a solid foundation for the development of antimicrobial strategies and therapeutics.
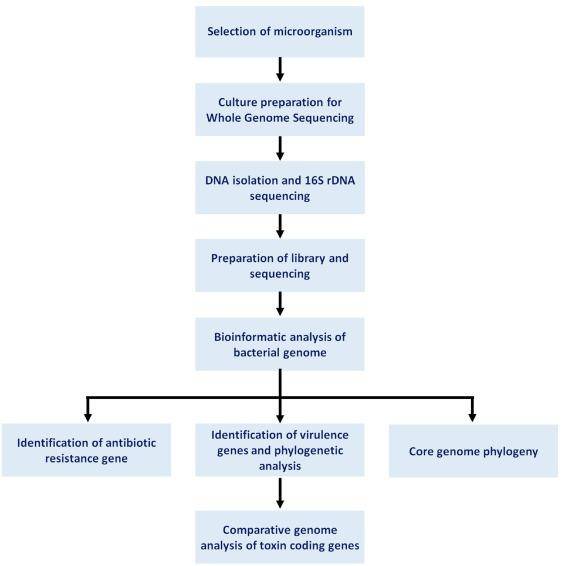 Workflow of demonstrating multiple virulence factors by sequencing. (Das et al., 2021)
Workflow of demonstrating multiple virulence factors by sequencing. (Das et al., 2021)
Conclusion
The identification of bacterial virulence genes stands as a pivotal research endeavor within the realms of bacteriology and biomedicine. Through the adoption of cutting-edge technologies and innovative methodologies, scientists are poised to delve deeper into the functions and mechanisms of virulence genes, providing invaluable support for antibacterial therapy and infection control. As technological advancements continue to unfurl, a profound understanding of bacterial virulence mechanisms promises to usher in groundbreaking discoveries in the realm of health and medical research.
Reference:
- Das, Soumya, et al. "Genome sequencing and annotation of multi-virulent Aeromonas veronii XhG1. 2 isolated from diseased Xiphophorus hellerii." Genomics 113.1 (2021): 991-998.


