
 Sample Submission Guidelines
Sample Submission Guidelines
Principles of Illumina Next-generation Sequencing (NGS)
The principal benefit of Illumina sequencing is its high throughput, which permits large-scale operation at low cost. Thanks to its high volume and cost-effectiveness, it can meet most sequencing needs and has heralded the era of high-throughput sequencing. Illumina sequencing has a vast range of applications. Initially utilized in genome assembly, it was later expanded to variation detection, RNA sequencing, single-cell sequencing, prenatal screening, tumor detection, and other fields.
Nonetheless, Illumina sequencing has a significant shortfall—its read length is relatively short. Progressing from an initial length of 35bp, it gradually increased to 75bp, 90bp, and currently the longest available, 2x300bp. Despite this, its read length remains somewhat short. This inadequacy can complicate the handling of repeat sequences and poses challenges for genome assembly, large fragment variation detection, and whole transcriptome research. The limitations inherent in Illumina technology constrain any further extension of read length, and this, to a certain extent, dampens its evolution.
The basic principle behind Illumina/Solexa Genome Analyzer sequencing is 'synthesis by sequencing'. During the process of DNA polymerase synthesizing the complementary strand, four different color-labeled dNTPs are consecutively added. The addition of each type of dNTP produces a specific fluorescent signal. This signal is captured and processed through specialized computer software, ultimately yielding the sequenced DNA data.
The workflow of Illumina sequencing can be broadly divided into four stages:
Library Construction
Cluster Generation
Sequencing
Base Recognition
Service you may interested in
Library Construction
What is DNA Library
To begin with, the concept of a 'library' needs to be elucidated. In this context, a library denotes a compilation of DNA fragments. In the process of library construction, sequence fragments undergo fragmentation, culminating in the formation of a DNA library.
In simple terms, it entails disrupting randomly ordered DNA molecules using techniques—such as sonication—into smaller fragments within a specific range of lengths. Unique adaptors are then appended at both ends of these minuscule segments, resulting in the construction of single-strand DNA libraries. These libraries are prepared and maintained for subsequent sequencing processes.
How to Construct a DNA Library
The initial step in library construction involves the random fragmentation of DNA samples. At this point, these DNA samples primarily consist of longer fragments, for instance, segments in the range of 100-300K. Through random fragmentation, these larger pieces are broken down into shorter fragments. There are diverse methods at our disposal for DNA fragmentation, including mechanical breakage, ultrasonication, and enzymatic digestion, among others.
The fragment length can be predetermined; for example, if we set the fragment length at 500 base pairs (bp), these larger pieces will eventually be broken into numerous 500bp short fragments, thus constructing a 500bp library. Other lengths can also be selected, such as the widely-used 170bp and 350bp libraries, as well as longer ones, including 500bp, 800bp, 2k, 5k, 6k libraries, and so forth. Typically, libraries with a size under 1000bp are referred to as short fragment libraries, while those with sizes larger are termed long fragment libraries.
It is crucial to note that, when referencing a 500bp library, 500bp serves merely as a peak value, indicating that the majority of fragment lengths approximate 500bp. Indeed, not all fragments measure precisely 500bp; there may be segments of varying lengths, such as 300bp or 800bp. Post-fragmentation, fragments within a certain range can be recovered through the electrophoresis process. For a 500bp library, fragments between 300-800bp can be recovered. The size of this library—or the insert size—is of monumental importance and will play a significant role in subsequent sequence assembly and short-read alignment processes. Once a suitable DNA library is retrieved, a series of subsequent procedures must be performed.
First, we add an adenine (A) base at the 3' end of the sequence. This conversion from a blunt end to a sticky end facilitates the connection to subsequent primers and adapters. After the addition of the adenine base, sequencing primers are integrated. Next, an index tag is introduced, a 6-8bp fragment, utilized to distinguish various sequencing samples. Given the massive amount of data generated in high-throughput sequencing technologies, such as 30G data from one lane, while sequencing a bacterial genome might only require 1G of data, it's possible to mix DNA from different species within a single sequencing process. Therefore, we must add unique index tags to establish distinction between mixed samples, be it animal, plant, or microbial DNA, for subsequent data partition.
Following index addition, termini adapter are incorporated. Adapter termini consist of the P7 and P5 termini, which are respectively integrated at both ends of the sequence. These P7 and P5 termini complementarily pair with the termini on the sequencing chip. Having undergone the procedures above, the sample can now be loaded onto the sequencing chip.
Cluster Generation
Following library construction, cluster generation is conducted – a critical step in the sequencing process. 'Cluster' refers to the amplification process of each DNA fragment. The aim of amplification is to strengthen the signal. During the sequencing process, it's necessary to excite the fluorescent groups of the bases and capture the corresponding fluorescent signals. With only one fluorescent group, its signal would be exceedingly weak. Therefore, through the enrichment process, the original single sequence is amplified into a cluster, allowing for signal magnification. A single fluorescent signal is weak and challenging to distinguish colour-wise, yet when put together in a cluster, the signal intensity augments significantly, making the fluorescence colours easier to discern.
The process of cluster generation takes place on a flowcell chip. The flowcell is a conduit for absorbing moving DNA fragments and serves as the core container for sequencing reactions, with all sequencing processes occurring here. When the library is prepared, the DNA can adhere randomly to the lanes of the flowcell surface.
Flowcell
 illumina sequencing flowcell (Image Source illumina)
illumina sequencing flowcell (Image Source illumina)
Within a given flow cell, there exist eight individual channels, herein referred to as eight 'lanes'. Each lane contains two chemically-modified surfaces—both top and bottom—abundantly seeded with primers, specifically P7 and P5 primers, which precisely pair with the adapters on the library. The rationale behind planting DNA onto a chip for sequencing emerges due to the constant fluid movement occurring during the sequencing process. Any DNA that fails to connect with the adapter is susceptible to detachment due to the fluid flow.
Each surface is divided into three 'swaths', with each swath containing 16 'tiles', which refer to small regions. Thus, a single lane contains 48 tiles (3 swaths multiplied by 16 tiles), summing the two surfaces to a total of 96 tiles. A full flow cell, therefore, encompasses 768 tiles (96 tiles multiplied by 8 lanes). As the chip becomes replete with sequencing adapters, the DNA capacity increases, leading to a corresponding surge in sequencing data volume. By injecting samples with appended primer adapters into the flow cell, the library is successfully planted on the chip.
Bridge PCR
In this step, we proceed to the bridge PCR amplification. To begin, the library is implanted onto the flowcell. During this process, a bridge PCR method is applied, which possesses certain distinctions from conventional PCR. In bridge PCR reactions, forward and reverse primers are both anchored to a flexible adaptor attached to the solid phase carrier (solid substrate). Subsequent to the PCR reaction, all amplification products from templates are immobilized at specific locations on the chip. As the junctions on both ends of the library complement the junction sequence base on the chip, upon the injection of samples into the flowcell, complementarity hybridization occurs, anchoring the library sequence to the chip.
dNTP and polymerase are subsequently added. The synthesis of a novel sequence from the primer along the template, complementary to the original sequence, is facilitated by the enzyme. Upon the addition of a concentrated sodium hydroxide solution, the DNA double strand unwinds into two single strands. While one strand combines with the adapter, the other strand does not and is flushed away as fluid passes. Neutral solvents and neutralizing solutions are subsequently added, causing the single-stranded DNA on the plate to undergo bending at one end and complementary hybridization with another primer on the chip.
We continue the addition of the polymerase and dNTP, facilitating PCR reactions, and synthesis of a new strand. This entire process is repeated with the addition of an alkaline solution and a neutralising one to ensure hybridization with a new adapter. What was originally a single strand has now become double stranded due to amplification. With several rounds of amplification, DNA quantity follows exponential growth, with the original singular strand eventually becoming a cluster of identical sequences, akin to a cloning process. This phenomenon is referred to as Bridge PCR, where an adapter at the DNA end hybridizes with an adapter on the chip, forming a curved 'bridge'. A single round of PCR amplification occurs on this 'bridge'.
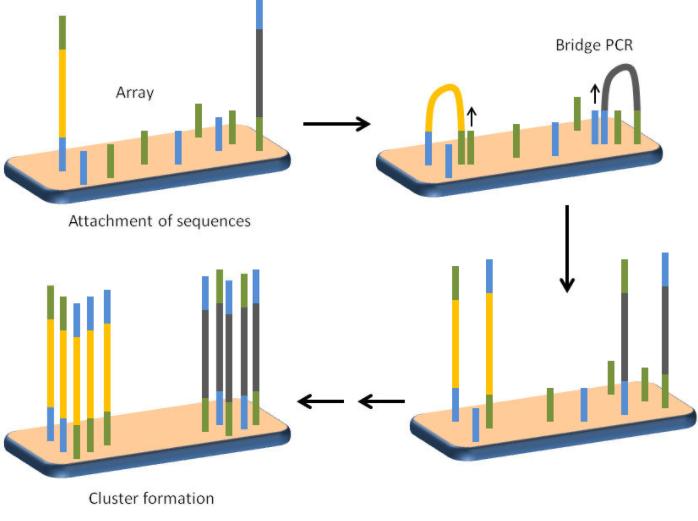 Schematic showing the concept of bridge PCR and cluster formation. (Sandeep Ameta 2013)
Schematic showing the concept of bridge PCR and cluster formation. (Sandeep Ameta 2013)
Sequencing
Once cluster generation is completed, sequencing can commence, employing the Illumina sequencing technology principle of concurrent sequencing by synthesis. The reaction system is simultaneously infused with DNA polymerase, adapter primers, and four types of fluorescently labeled dNTPs (similar to Sanger sequencing). The 3'-OH group of these dNTPs is chemically protected, ensuring that only one dNTP can be incorporated at a time, thus, only one base is added during each sequencing step.
After a dNTP is added to the synthesizing chain, unused free dNTPs and DNA polymerase are rinsed off. Next, a buffer is added to induce fluorescence, which is excited by laser light and recorded using an optical apparatus. Finally, computer analysis converts the optical signal to a sequencing base.
After recording the fluorescence, a chemical reagent is introduced to quench the fluorescent signal and remove the protecting group from the 3'-OH of the dNTP, preparing for the next round of sequencing reactions. This process enhances the quality of the sequencing, increasing data output, and ensuring precision.
Upon completion of a sequencing round, both the fluorescent group and the 3' terminus blocking group are excised. This step is intended to eliminate these groups, thus allowing for the continuation of synthetic reactions, a unique feature of reversible terminator blocking technology. Subsequently, new dNTPs and synthesis enzymes are added with the aim of constructing new nucleotides. Following exposure to stimulated light, fluorescent signals are captured and analyzed, hence identifying the second nucleotide. This process is continually repeated, resulting in an increasing number of nucleotides being sequenced and thereby extending the length of the sequence. The sequencing continues until termination and the results of the single-strand sequencing are subsequently presented.
Illumina sequencing technology adopts paired-end sequencing, where both the forward and reverse strands undergo a sequencing process. First, a synthesis is conducted, leading to the creation of a double strand - namely, the complementary strand of the original sequencing strand. The original strand is then excised using chemical reagents, leaving behind only the complementary strand. Based upon this configuration, the sequencing process proceeds. Likewise, a method of simultaneous synthesis and sequencing is utilized, where nucleotide synthesis, fluorescent group excitation, fluorescent signal capture, and fluorescent group plus 3' terminus blocking group excision occur. Following this process, the next round of synthesis sequencing is conducted. This cycle continues until all sequencing tasks are completed.
Sequencing by Synthesis
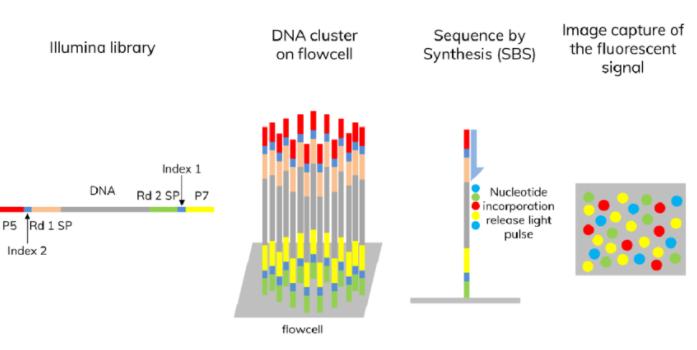 Illumina sequencing by synthesis (SBS) (Syahzuwan Hassan et al,. 2023)
Illumina sequencing by synthesis (SBS) (Syahzuwan Hassan et al,. 2023)
With regards to the distinct nature of Illumina sequencing technology, it uniquely adds only one dNTP at a time. It effectively resolves the challenge of measuring the lengths of homopolymers. Its primary sequencing errors originate from base substitutions, with current error rates ranging approximately from 1% to 1.5%. To provide a case in point, for human genome resequencing, Hisq series with a 30x to 50x sequencing depth would require between 3 to 5 days. Comparatively, the recently introduced NovaSeq series would only need approximately 40 hours.
Illumina Sequencing volume comparison
| Sequencer | Total Data Output per Sequencing | Number of Reads (Billion) | Sequencing Read Length (bp) | Sequencing Time |
| HiSeq 2500 | 720-800 Gb | 8.0 | PE 100 | 5 days |
| HiSeq 4000 | 1,500 Gb | 10.0 | PE 150 | 3.5 days |
| NovaSeq 5000 | 850-1,000 Gb | 2.8-3.3 | PE 150 | 1.7 days |
| NovaSeq 6000 | 3,000 Gb | 10.0 | PE 150 | 1.7 days |
According to data volume estimates, a NovaSeq6000 (S4) sequencer running at full capacity can complete sequencing for over 6400 individuals annually. Notably, the data publicized by Illumina is typically conservative. In practical application, we have found that the proportion of high-quality (Q30) read segments exceeds 90% of the total data, significantly higher than the 75% officially announced. Therefore, the actual total data yield is also higher than expected.
Base Recognition
Following the completion of sequencing, we obtain a multitude of fluorescence signal files, rather than IMMEDIATE sequences of adenine (A), thymine (T), cytosine (C), and guanine (G) bases. These files have to undergo image processing, transforming them into color-coded spot files, which are then stored in bcl format. The extraction process from these bcl files to retrieve the bases is termed as basecalling. Each spot file documents an array of information, inclusive of the lane number, tile number, x and y-axis coordinate positions of the spots, and the light intensity of each A, T, C, and G cycle. However, the bcl files are in a binary format, which does not match the fastq format text file we ultimately require. Therefore, it necessitates the utilisation of a bcl2fastq conversion software to transform the bcl files appropriately.
Every image corresponds to a photo captured during a sequencing. We can easily differentiate between red, yellow, green, and blue, with each color representing a different type of base. The image from the second sequencing reads the second base, always from the exact same location. Reading from the same position during each sequencing culminates in constructing a sequence. Essentially, this process involves joining adjacent images and extracting base groups from the same location, thereby establishing a sequence.
The aforementioned description of determining the type of base according to the color of the image is merely a simplification. The actual situation is significantly more complex. Among the four bases, purines and pyrimidines have similar chemical structures, and the wavelengths of the four fluorescent base groups overlap. Therefore, the type of base cannot be immediately discerned by color alone, especially under non-cluster circumstances, where judgments are even more challenging.
As a matter of fact, the sequencer determines the identification by analyzing the contribution rate of the four fluorescent materials at four different wavelengths. For example, as the table shows, the contribution rates of the four fluorescent materials to the four wavelengths are distinctive, forming a four-dimensional quaternion contribution rate matrix. Hence, when identifying each illumination point, it's akin to solving a set of quaternary linear equations. The identification of this base corresponding to the illuminated point is made by choosing the one with the highest probability. This complex process can be automatically executed via the built-in software of the sequencer. Ultimately, the generated fastq sequence file is the desired sequencing data.
FAQ of Illumina Next-generation Sequencing
Q: In achieving repetitive experiments, why opt for repetitive addition of sodium hydroxide and neutral solutions rather than directly utilizing the variable temperature operation principle of PCR machines?
A: The repetitive addition of sodium hydroxide and neutral solutions serves the purpose of removing and recovering DNA fragments, which aids in the preparation of samples for sequencing. This is primarily because the Illumina sequencing technology employs the "Bridge PCR" technique rather than the traditional liquid-phase PCR. Bridge PCR is a critical operation within the Illumina sequencing process, involving the fixation of PCR products onto the surface of the sequencing chip to form DNA "bridges," which subsequently undergoes sequencing reactions. Bridge PCR requires a substantial amount of DNA fragments to be bound to the surface, as opposed to DNA amplification in a liquid phase. Consequently, direct use of the liquid phase PCR principle of PCR machines fails to facilitate this fixation of DNA fragments onto the chip surface.
Q: Are clustered sequences identical? And what if more than one DNA fragment binds before amplification?
A: Typically, in a cluster sequence, each DNA fragment shares the same sequence. Nevertheless, during sequencing, there might be instances in which more than one DNA fragment binds initially, an occurrence referred to as "cluster overlap" or "cluster coalescence". Cluster overlap can arise during DNA library creation or PCR amplification processes where some DNA fragments cluster together and are immobilized on the chip's surface within the same sequence cluster.
Cluster overlap can potentially instigate problems with sequencing data such as:
Superimposed signals: During sequencing, the presence of more than one DNA fragment in a singular sequence cluster could lead to overlapping signals, which can interfere with sequencing outcomes.
Low-quality data: If an abundance of DNA fragments are present within a singular sequence cluster, it could decrease the effective sequencing rate for each fragment, thereby hampering data quality.
To minimize cluster overlap, control measures regarding fragment concentration and quantity are often applied during the DNA library creation and PCR amplification processes, ensuring only one DNA fragment binds within each sequence cluster. Furthermore, the Illumina sequencing platform resorts to a series of image processing and data analysis algorithms to eliminate or correct interference signals caused by cluster overlap, thereby enhancing data quality and accuracy.
Q: Within Illumina sequencing, do all the DNA fragments within a cluster produce identical fluorescent signals? During sequencing, is there a possibility of one strand still being dyed yellow while an adjacent strand has finished yellow coloring and started shifting to blue?
A: Generally, all DNA fragments within a given cluster yield homogeneous fluorescent signals in an Illumina sequencing run. Concerning the pace of this process, it's essential to note that sequencing usually transpires concurrently or at a very similar rate across strands. Hence, situations where one DNA strand lags in dyeing while another strand has advanced onto a different nucleotide base are unlikely.
However, it is not an absolute scenario. Practical complications might lead to sequencing errors or stray signals. Additionally, sequencing reliability can degrade over time—speed non-uniformities can develop, degrading the sequencing quality further downstream the fragments. To counteract such an issue, we employ paired-end (or double-ended) sequencing approach. This method enhances sequence resolution, particularly for long-read sequences, ensuring a more nuanced understanding of the process.
Q: Why is the number of cycles set in relation to the length of the reads?
A: During the sequencing process, DNA samples are subjected to enzymatic amplification and subsequent sequencing reactions. To achieve the required read length, appropriate numbers of amplification and sequencing cycles must be performed to ensure that we have covered an adequate number of base pairs. Therefore, we consider the number of cycles to be intimately linked with the read length requirement. Put another way, if a read of a specific length is required, the sequencer adjusts the number of cycles to meet this read length target. Hence, the number of cycles is a parameter set to fulfill the requirement for read length.
Q: Why is it necessary to utilize paired-end sequencing?
A: In paired-end sequencing, one end of the DNA fragment is initially sequenced, followed by a secondary sequencing of the other end, leading to two distinct sets of sequencing data. It must be noted that as the sequencing process advances, the sequencing quality tends to deteriorate. For instance, toward the downstream phase of single-end sequencing, the quality often falls short of the desired precision. Implementing paired-end sequencing allows simultaneous sequencing of both ends of a DNA fragment, ensuring a notably superior sequencing quality at the upstream end. This, when coupled with the assembly of the two sequencing outputs, significantly enhances the overall quality of sequencing, consequently extending the effective length of the sequencing read.


