
 Sample Submission Guidelines
Sample Submission Guidelines
miRNA Sequencing Workflow: A Comprehensive Overview
miRNAs are integral to the regulation of gene expression post-transcriptionally, orchestrating diverse cellular processes fundamental to organismal development, homeostasis, and disease pathogenesis. The advent of next-generation sequencing (NGS) has revolutionized miRNA research, offering unprecedented insights into the complex landscape of small RNA expression. A comprehensive knowledge of their regulatory functions and expression patterns is pivotal to deciphering their respective roles in health and pathological states. miRNA sequencing, a formidable technique, facilitates a thorough profiling of miRNA expression, thus shedding light on their functional implications. In this discourse, we present an elaborate overview of the miRNA sequencing workflow and protocol, underscoring each constituent step in the process.
How Does miRNA Sequencing Work
1. Sample Preparation
The miRNA sequencing process commences with sample preparation, wherein high-quality RNA is extracted from the pertinent biological specimens. Diverse sample sources, ranging from tissues, cells, biofluids, to environmental samples, are amenable for miRNA sequencing analysis. Extraction methodologies for total RNA are chosen judiciously to ensure the preservation of small RNA species, with commonly utilized techniques including TRIzol extraction or column-based purification kits. Rigorous attention is devoted to mitigating RNA degradation and contamination risks throughout the handling and processing phases.
2. RNA Quality Control
Following RNA extraction, the quality and quantity of the extracted RNA are evaluated employing spectrophotometric methods, such as UV absorbance, and electrophoretic techniques like agarose gel electrophoresis. Maintaining RNA integrity is paramount for successful miRNA sequencing, as degraded RNA can compromise the accuracy and robustness of sequencing outcomes. Consequently, stringent criteria are applied to select high-quality RNA specimens with preserved small RNA fractions for subsequent library construction.
3. Small RNA Library Preparation
The next step in the miRNA sequencing workflow is the generation of small RNA libraries. Small RNA molecules, including miRNAs, are enriched from the total RNA pool using size selection methods, such as gel electrophoresis or size-exclusion chromatography. The enriched small RNA fraction is then ligated with adapters that facilitate reverse transcription and amplification. This step introduces unique barcodes or indexes to individual samples, allowing for multiplexed sequencing of multiple samples in a single sequencing run.
4. Reverse Transcription and PCR Amplification
Upon ligation, a miRNA-specific primer is employed to convert the small RNAs adhered to adapters into complementary DNAs (cDNAs). At this point, a polymerase chain reaction (PCR) is used to accentuate the cDNA library and integrate suites necessary for downstream sequencing. Meticulous streamlining of PCR conditions is pivotal to minimalize bias and reduce the probability of artifacts during amplification, thereby guaranteeing the alignment of the the miRNA abundance in the final sequencing library with the original sample.
5. Library Quality Control
After PCR amplification, the quality and quantity of the small RNA libraries are assessed utilizing analytical methodologies such as capillary electrophoresis (e.g., Agilent Bioanalyzer) or fluorometric techniques (e.g., Qubit fluorometer). Evaluation of library size distribution and concentration is imperative to ascertain adherence to sequencing criteria. Subsequently, libraries that successfully meet quality control standards undergo normalization and pooling in preparation for sequencing.
6. Sequencing
The combined small RNA libraries are introduced onto a next-generation sequencing platform, such as Illumina MiSeq, HiSeq, or NovaSeq, to undergo sequencing-by-synthesis. Throughout this process, fluorescently labeled nucleotides are successively incorporated into complementary DNA strands, yielding sequence reads that faithfully depict the small RNA composition of the specimen. The sequencing depth, quantified by the number of reads generated per sample, is tailored to meet the desired coverage and sensitivity necessary for precise miRNA expression analysis.
7. Data Preprocessing and Quality Control
After sequencing, the raw data produced by the sequencer undergo preprocessing to eliminate adapter sequences, low-quality reads, and sequencing artifacts. Quality control parameters, such as per-base sequence quality and GC content distribution, are scrutinized to uphold data integrity. Preprocessing procedures may additionally involve the excision of low-quality bases and the culling of reads according to length and sequence composition criteria.
8. Alignment and Mapping
The preprocessed sequencing reads are aligned to reference genomes or miRNA sequence databases utilizing specialized alignment algorithms, such as Bowtie, BWA, or miRDeep2. In instances where species boast well-annotated genomes, reads can be directly mapped to genomic coordinates corresponding to established miRNAs. Conversely, when reference genomes are absent or incomplete, reads may be aligned to miRNA sequence databases to discern known miRNAs or employ de novo miRNA prediction methodologies.
9. Quantification of miRNA Expression
Upon mapping the reads, the expression levels of miRNAs are quantified by tallying the number of reads aligning to each miRNA sequence. To mitigate discrepancies stemming from variations in sequencing depth and library size across samples, normalization techniques such as reads per million (RPM) or transcripts per million (TPM) are employed. The resultant miRNA expression profiles furnish quantitative insights into the abundance of individual miRNAs within the samples.
10. Differential Expression Analysis
Statistical methodologies are employed to discern differentially expressed miRNAs amidst varying experimental conditions or sample cohorts. Differential expression analysis entails a comparative assessment of miRNA expression levels across samples, meticulously considering factors such as biological replicates and experimental design nuances. Significantly dysregulated miRNAs are pinpointed through rigorous statistical criteria encompassing fold change and adjusted p-values, thereby elucidating miRNA-mediated regulatory shifts linked to biological processes or pathological conditions.
11. Functional Annotation and Pathway Analysis
Differential expression of miRNAs prompts a rigorous examination to unveil their functional significance within biological frameworks, encompassing cellular pathways and disease etiologies. This exploration necessitates a multifaceted approach, integrating miRNA expression profiles with cutting-edge bioinformatics resources and databases. Through this integration, we undertake pathway enrichment analyses, forecast target gene candidates, and delineate regulatory networks modulated by aberrant miRNA expression patterns. By delving into functional annotation and pathway analyses, we gain profound insights into the pivotal roles of miRNA expression alterations, thereby unraveling their intricate involvement in disease pathophysiology.
12. Validation and Experimental Validation
In conclusion, following the identification of prospective miRNA candidates via sequencing analysis, rigorous validation ensues through experimental methodologies such as quantitative real-time polymerase chain reaction (qRT-PCR), Northern blotting, or functional assays. These validation procedures serve to corroborate the expression profiles and regulatory impacts of the identified miRNA candidates, thus solidifying the outcomes derived from miRNA sequencing analyses. Such experimental validation stands as a cornerstone, essential for affirming the credibility and biological significance of miRNA sequencing outcomes, while also furnishing valuable functional elucidations regarding the involvement of miRNAs within distinct biological milieus.
13. Data Interpretation and Biological Insights
The outcomes derived from miRNA sequencing analysis, in conjunction with experimental validation data, undergo meticulous interpretation aimed at elucidating the intricate regulatory functions of miRNAs in both physiological well-being and pathological states. Through an integrative approach, wherein miRNA expression profiles are harmonized with diverse omics datasets encompassing mRNA expression, proteomics, and epigenomics, a holistic comprehension of miRNA-driven regulatory networks is attained, shedding light on their profound influence on cellular dynamics and disease manifestations. This interpretive endeavor not only fosters hypothesis generation and prompts the formulation of pertinent research inquiries but also unveils potential therapeutic targets warranting further exploration and scrutiny.
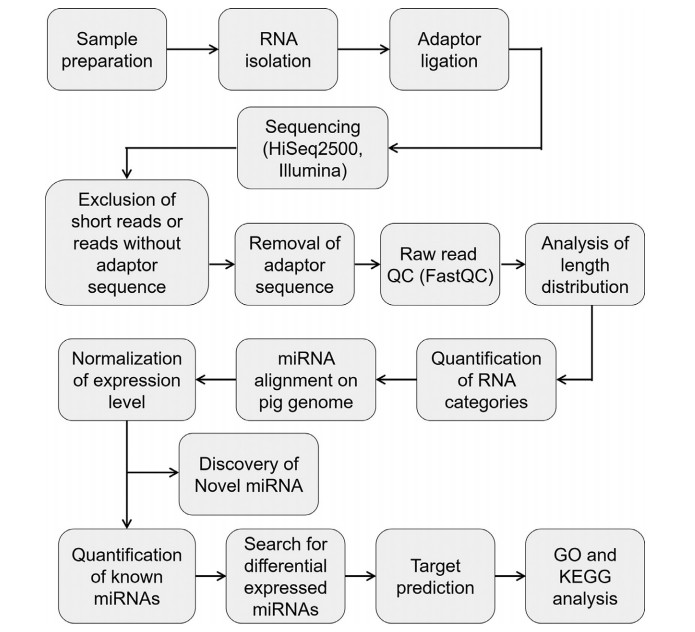 Workflow of miRNA-sequencing and data analysis.
Workflow of miRNA-sequencing and data analysis.
Service you may intersted in
Learn more
Complete Overview of Small RNA Sequencing: Methods, Workflow, Platform, and Applications
miRNA Sequencing Protocol
In this continuation of the miRNA sequencing workflow, we will delve into the protocol detailing the steps involved in miRNA library preparation, sequencing, and data analysis.
miRNA Library Preparation Protocol
Materials:
Total RNA samples
Small RNA library preparation kit (e.g., CD Small RNA Library Prep Set for Illumina)
Nuclease-free water
Adapters and primers
RNase inhibitor
Reverse transcriptase
PCR reagents (e.g., Taq polymerase, dNTPs, PCR buffers)
Procedure:
Adapter Ligation: Mix RNA samples with 3' and 5' RNA adapters and ligase buffer. Incubate the mixture at an appropriate temperature for adapter ligation.
Reverse Transcription: Add reverse transcriptase and RNase inhibitor to the ligated RNA samples to generate cDNA. Incubate the reaction mixture at an appropriate temperature for reverse transcription.
PCR Amplification: Amplify cDNA using PCR primers that anneal to the adapters. Perform PCR under optimized conditions to amplify the small RNA library. Incorporate barcode sequences for multiplexing if desired.
Library Cleanup: Purify amplified libraries using size selection methods (e.g., gel electrophoresis or magnetic bead-based purification) to remove unincorporated adapters and primer dimers.
Library Quality Control: Assess library quality and quantity using analytical techniques such as capillary electrophoresis or fluorometric quantification. Validate library size distribution and concentration before pooling for sequencing.
Sequencing Protocol
Materials:
Small RNA libraries
Sequencing platform (e.g., Illumina MiSeq, HiSeq, or NovaSeq)
Sequencing reagents (e.g., flow cells, sequencing primers, and buffers)
Indexing reagents for multiplexed sequencing
Procedure:
Library Denaturation: Denature small RNA libraries to generate single-stranded DNA templates for sequencing cluster generation.
Cluster Generation: Immobilize denatured libraries onto the sequencing flow cell surface using bridge amplification to generate clusters of identical DNA fragments.
Sequencing-by-Synthesis: Perform sequencing-by-synthesis using fluorescently labeled nucleotides to generate sequence reads from the DNA clusters. Monitor fluorescence signals to determine the sequence of incorporated nucleotides.
Base Calling: Convert fluorescence signals into nucleotide base calls using sequencing software provided by the sequencing platform.
Data Generation: Collect raw sequencing data, including sequence reads and quality scores, from the sequencer for downstream analysis.
Data Analysis Protocol
Bioinformatics Pipeline:
Preprocessing: Trim adapter sequences and low-quality bases from raw sequencing reads. Filter out short reads (< 15 nucleotides) and sequences with low complexity.
Alignment: Map processed reads to reference genomes or miRNA sequence databases using alignment algorithms (e.g., Bowtie or BWA).
Quantification: Count the number of reads that align to each miRNA sequence. Normalize read counts to correct for differences in sequencing depth and library size.
Differential Expression Analysis: Identify differentially expressed miRNAs between experimental conditions using statistical methods (e.g., DESeq2 or edgeR).
Functional Annotation: Functionally annotate miRNAs based on target gene prediction, pathway enrichment analysis, and regulatory network analysis.
Validation: Validate sequencing results using experimental techniques such as qRT-PCR or functional assays to confirm the expression patterns and regulatory effects of candidate miRNAs.
Conclusion
The miRNA sequencing workflow provides a robust framework for profiling miRNA expression and elucidating their functional significance in biological processes and disease. By following the protocol outlined above, researchers can generate high-quality miRNA sequencing data, analyze differential expression patterns, and gain insights into miRNA-mediated regulatory networks. This comprehensive approach facilitates the discovery of novel biomarkers, therapeutic targets, and mechanistic insights into miRNA biology, contributing to advancements in biomedical research and precision medicine.
References:
- Haseeb, A., Makki, M.S., Khan, N.M. et al. Deep sequencing and analyses of miRNAs, isomiRs and miRNA induced silencing complex (miRISC)-associated miRNome in primary human chondrocytes. Sci Rep 7, 15178 (2017).
- Jardillier R, Koca D, Chatelain F, Guyon L. Optimal microRNA Sequencing Depth to Predict Cancer Patient Survival with Random Forest and Cox Models. Genes (Basel). 2022
- Campbell JD, Liu G, Luo L, Xiao J, Gerrein J, Juan-Guardela B, Tedrow J, Alekseyev YO, Yang IV, Correll M, Geraci M, Quackenbush J, Sciurba F, Schwartz DA, Kaminski N, Johnson WE, Monti S, Spira A, Beane J, Lenburg ME. Assessment of microRNA differential expression and detection in multiplexed small RNA sequencing data. RNA. 2015
- Khamina K, Diendorfer AB, Skalicky S, Weigl M, Pultar M, Krammer TL, Fournier CA, Schofield AL, Otto C, Smith AT, Buchtele N, Schoergenhofer C, Jilma B, Frank BJH, Hofstaetter JG, Grillari R, Grillari J, Ruprecht K, Goldring CE, Rehrauer H, Glaab WE, Hackl M. A MicroRNA Next-Generation-Sequencing Discovery Assay (miND) for Genome-Scale Analysis and Absolute Quantitation of Circulating MicroRNA Biomarkers. Int J Mol Sci. 2022
- Potla P, Ali SA, Kapoor M. A bioinformatics approach to microRNA-sequencing analysis. Osteoarthr Cartil Open. 2020 Dec 19;3(1):100131. doi: 10.1016/j.ocarto.2020
- Pérez-Rodríguez D, López-Fernández H, Agís-Balboa RC. Application of miRNA-seq in neuropsychiatry: A methodological perspective. Comput Biol Med. 2021 Aug;135:104603. doi: 10.1016/j.compbiomed.2021
- Oh JN, Son D, Choi KH, Hwang JY, Lee DK, Kim SH, Lee M, Jeong J, Choe GC, Lee CK. MicroRNA expression data of pluripotent and somatic cells and identification of cell type-specific MicroRNAs in pigs. Data Brief. 2020
- Motameny S, Wolters S, Nürnberg P, Schumacher B. Next Generation Sequencing of miRNAs - Strategies, Resources and Methods. Genes (Basel). 2010


