
 Sample Submission Guidelines
Sample Submission Guidelines
Detection methods for Genetic Mutations Analysis
As a focal point in current life science research, genetic mutation enjoys rapidly evolving detection methodologies synonymous with the field. The advancements in genetic mutation detection techniques hold significant meaning for early disease diagnosis and treatment. The aim of this paper is to provide a concise exposition of various commonly used genetic mutation detection methodologies and their underlying principles. This is intended to aid in appropriate selection based on testing objectives and experimental conditions.
Southern blot
Conceived by the eminent British biologist Edwin Southern in 1975, Southern blot hybridization is a pioneering technique developed for the specific detection of DNA. Anchored in the fundamental principle of base complementarity, this method exploits the precise and selective hybridization of two nucleic acid single strands exhibiting homology under certain conditions, leading to the formation of a double-stranded structure. As the first embodiment of blotting methodologies, Southern blot hybridization maintains a vital presence in the field of molecular biology.
At its core, the mechanism of Southern blot hybridization is founded on the principle of complementary base pairing. This process involves nucleic acid strands, which, possessing a certain degree of homology, selectively participate in the formation of double-stranded structures under predetermined conditions. Provided that the target material incorporates sequences complementary to the probe, their interaction is enabled through precise base pairing. Upon the removal of unbound probes through washing processes, the identification and quantification of the hybridized fragments are achieved using modalities such as autoradiography (Figure 1) or other apposite methodologies.
In light of its superior sensitivity and specificity, Southern blot hybridization rapidly gained recognition as the archetypal molecular detection method in the spectrum of probe hybridization techniques. Its utility spans across myriad fields, such as the delineation of gene mutations and the scrutiny of Restriction Fragment Length Polymorphism (RFLP).
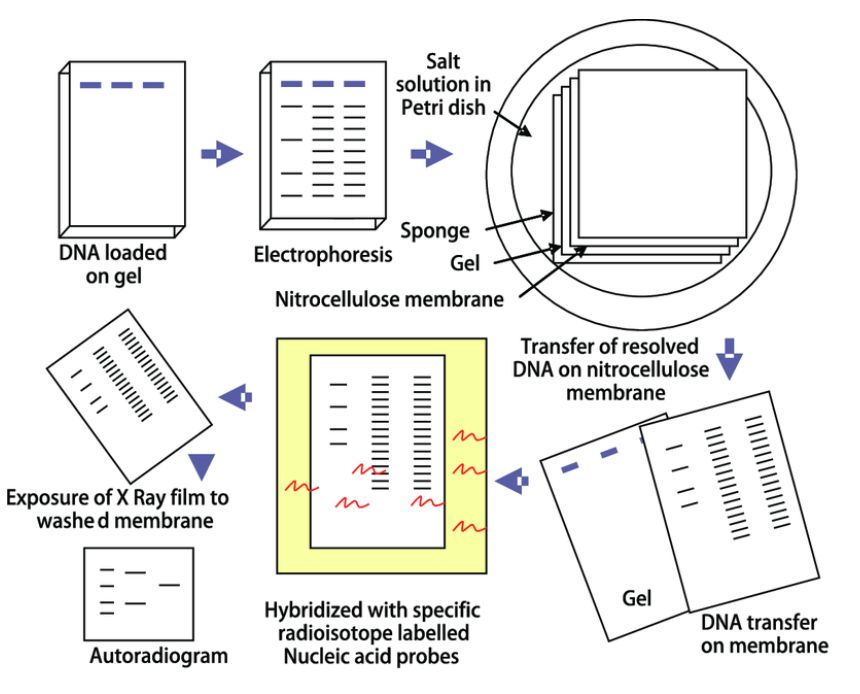 Figure.1 Southern blot analysis (Rajiv Kumar et al.,2012).
Figure.1 Southern blot analysis (Rajiv Kumar et al.,2012).
PCR
In the domain of gene mutation detection, Southern blot technology has held a prominent position since 1985, capable of discerning various mutation forms such as gene deletions, insertions, and frameshift recombinations. However, under the technological constraints of that era, the inability to artificially amplify target genes within samples rendered certain mutations undetectable by Southern blot. Consequently, mutations eluding Southern blot scrutiny necessitated intricate in vitro oligonucleotide-mediated DNA mutagenesis, followed by laborious DNA sequence analysis for confirmation—a method characterized by its complexity, time-consuming nature, and elevated costs.
The inception of Polymerase Chain Reaction (PCR) technology heralded a paradigm shift in molecular diagnostics, particularly concerning gene mutation detection. Essentially, all modern molecular diagnostic methods pivoted around the PCR methodology. The implementation of PCR technology effectuated substantial advancements in multiple dimensions of detection: it automatized procedures, condensed the timescale of analyses, and significantly bolstered result precision. By all means, the integration of PCR technology constituted a seminal development in gene mutation detection research.
1. ARMS-PCR
The Amplification Refractory Mutation System- Polymerase Chain Reaction (ARMS-PCR), also known as Allele-Specific PCR (AS-PCR), rests upon the principle of allele-specific extension reaction. This technique only proceeds when a specific allele's primer pairs align with the mutation site, allowing the extension reaction to occur. Globally, ARMS-PCR has emerged as one of the most critical and advanced technologies within the realm of individual molecular detection for tumors. Simultaneously, it stands as one of the most accurate technological instruments in the field of genetic disease detection.
The distinction between ARMS-PCR and conventional PCR lies in the design of the primer for allele-specific genes. Two upstream primers have differing 3' end sequences. During the PCR amplification process, upstream primers that cannot fully match the template fail to form completely complementary base pairs, leading to mismatching. Once this mismatching reaches a certain level, primer extension is terminated, and no PCR amplification product of specific length can be obtained, indicating that the template DNA does not have a base pair with the 3' end of the primer, and vice versa (Peng et al.,2018). At present, scientists have extended the ARMS-PCR methodology to incorporate four primers (Tetra-Primer ARMS-PCR), whereby four PCR primers are designed simultaneously, two of which are located on the 3' end SNP site and have opposite directions belonging to different genotypes, while the other two are located outside the SNP (Figure 2).
Service you may intersted in
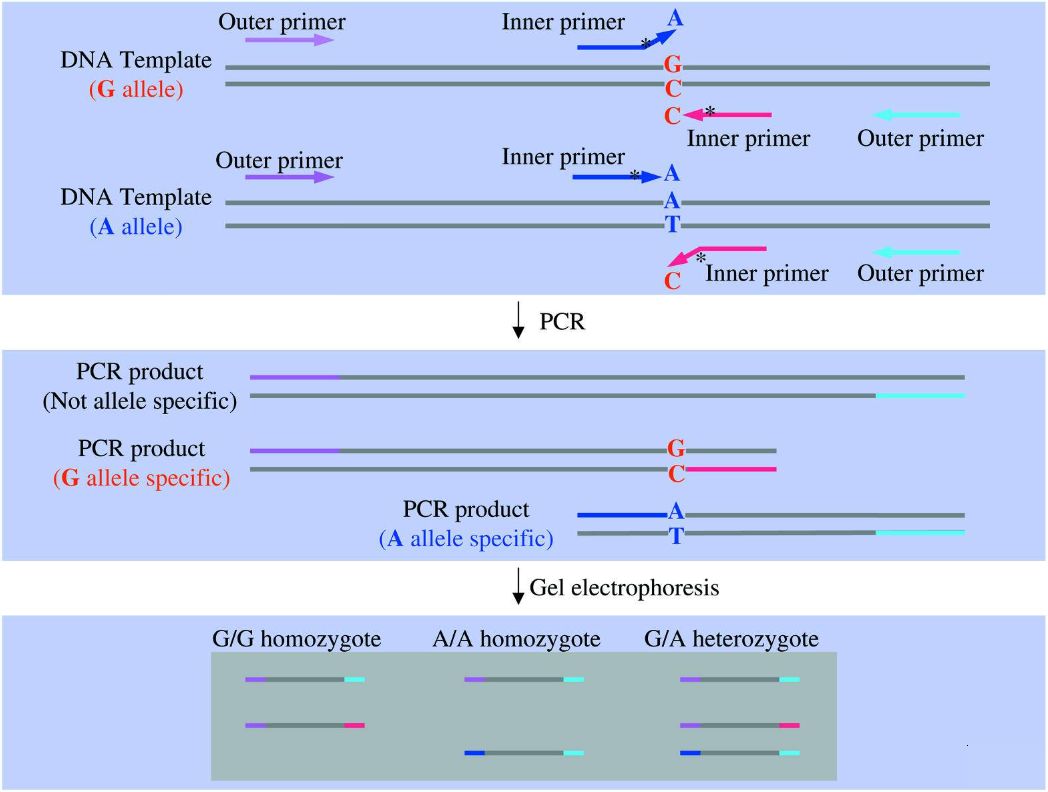 Figure.2 ARMS-PCR (Ye et al., 2001)
Figure.2 ARMS-PCR (Ye et al., 2001)
2. Digital PCR
Digital PCR (dPCR) epitomizes an advanced technique for the absolute quantification of nucleic acids. Heralded as the third generation of PCR technologies, it is considered seminal in the arena of life sciences and clinical diagnostics. This concept was first brought to light in 1999 by luminaries Bert Vogelstein and Kenneth W. Kinzler.
Digital PCR is a unique system that micropartitions the samples into droplets, ensuring an average of one or zero target DNA molecules in each partition. Post PCR amplification, the presence or absence of fluorescence in discrete partitions is recorded using a binary system. Under this system, the presence of fluorescence is denoted as 1 and non-fluorescence as 0. Subsequent statistical analysis of the fluorescence signals yields an accurate absolute quantification of wild-type and mutant species within the testing sample. This high level of sensitivity allows for the detection of mutations as low as a single copy. (Figure 3)
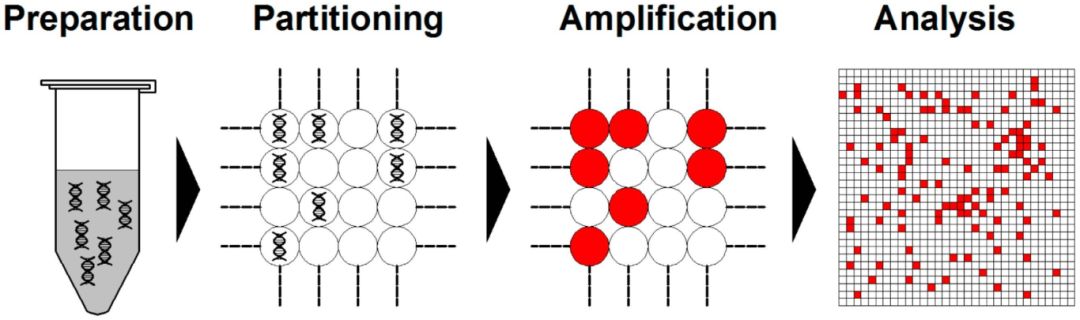 Figure.3 dPCR workflow (Cao et al., 2020)
Figure.3 dPCR workflow (Cao et al., 2020)
3. HRM
High-Resolution Melting Curve (HRM) analysis, invented by Professor Carl Wittwer of Molecular Pathology at the University of Utah, is a technological advancement predicated on polymerase chain reaction (PCR); it's specially designed for gene mutation detection and genotyping. HRM primarily hinges on a system that faithfully combines PCR target fragments aptly designed for the purpose of use, a saturating DNA double-strand fluorescent dye, an instrument system capable of thermally-regulated high-resolution melting curve signal attainment, and a melting curve software analysis system. The technique, owing to its simplicity, cost-effectiveness, speed, closed-tube post-PCR mode that mitigates the risk of secondary contamination, and high detection sensitivity, has rapidly become widely adopted for various gene analyses in medical and other bio-related fields.
The main principle of HRM exploits the property of a saturating type of double-stranded DNA (dsDNA) dye capable of integrating into the PCR product. It involves real-time monitoring of dsDNA unwinding during the heating process, the subsequent detachments of the fluorescent dye leading to a reduction or total disappearance of fluorescent signals, and accurately recording the melting curve at high resolution. Thereby, allowing for a highly sensitive analysis of sample mutations, single nucleotide polymorphisms (SNPs), and methylation sites at the resolution of a single base.
Service you may intersted in
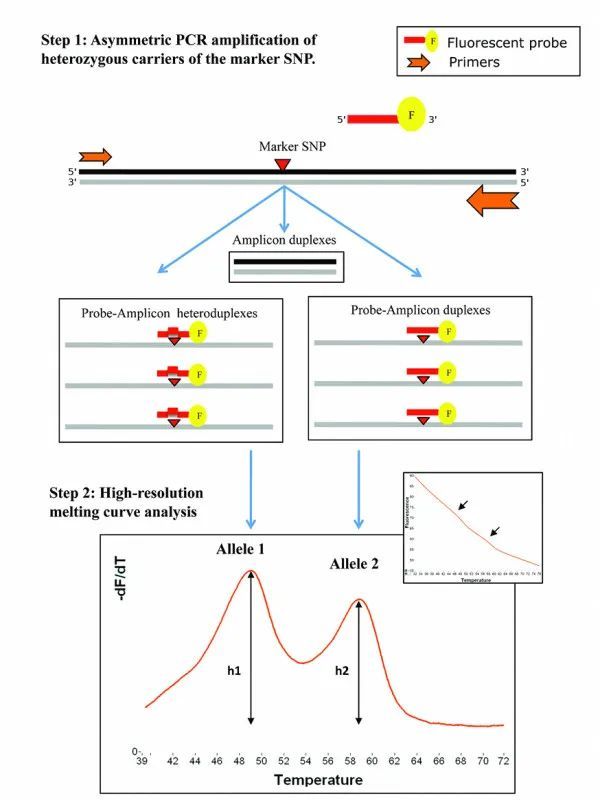 Figure.4 HRM principle(Nguyen-Dumont et al., 2011)
Figure.4 HRM principle(Nguyen-Dumont et al., 2011)
The limitations of HRM technology, similar to other methods for detecting gene mutations, include difficulties in identifying unknown mutation sites and its sensitivity being influenced by the type of Single Nucleotide Variants (SNVs) in the genetic segment under examination. However, akin to other gene testing methods that target specific gene fragments, HRM technology offers several merits including flexibility, speed, high sensitivity, and cost-effectiveness. Furthermore, it has the capacity to detect all types of gene mutations. It also offers the adaptability to increase the testing throughput, which conserves the amount of sample used, reduces the testing time and cost, and minimally potentiates the risk of contamination — an optimum solution to meet the demands of clinical testing.
4. IAT
Isothermal Amplification Technology (IAT) is another method that evolved from traditional PCR. It operates at a constant temperature to amplify DNA, rendering it exceedingly rapid, sensitive, straightforward, portable, and highly specific. IAT holds outstanding application potential within molecular diagnostics, particularly point-of-care testing and on-site screening. The foundational principle of IAT resides in using various enzymes, such as strand-displacement polymerases, RNA spliceosomes etcetera, to complete DNA amplification under isothermal conditions based on a circular DNA amplification scheme. During IAT, appropriate primer design can yield different lengths of RNA fragments through the function of an RNA spliceosome. The detection of these RNA fragments facilitates analysis of specific mutations within the DNA sequence.
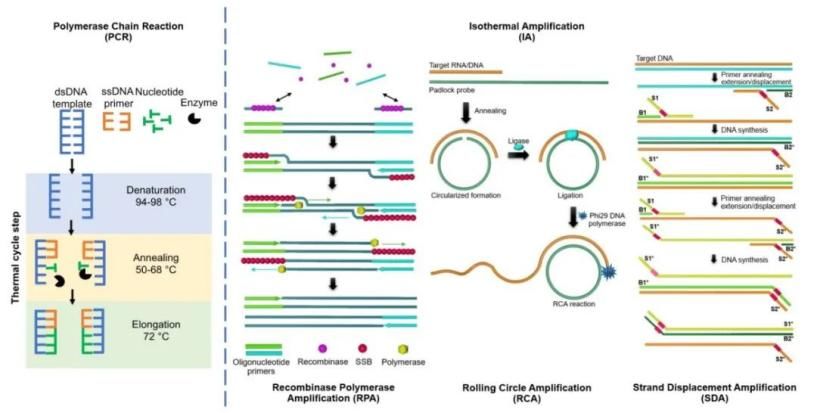 Figure.5 PCR and some IAT systems(Boonbanjong et al., 2022)
Figure.5 PCR and some IAT systems(Boonbanjong et al., 2022)
With the continual advancement of biotechnological methodologies, IAT have given rise to a diverse array of techniques. These include Loop-mediated Isothermal Amplification (LAMP), Rolling Circle Amplification (RCA), Nucleic Acid Sequence-Based Amplification (NASBA), Helicase-Dependent Isothermal DNA Amplification (HDA), Recombinase Polymerase Amplification (RPA), Nicking Enzyme Amplification Reaction (NEAR), Strand Displacement Amplification (SDA), and Hybridization Chain Reaction (HCR).
Gene sequencing
The essence of gene sequencing involves utilizing a gene sequencer to determine the arrangement of the four nucleobases (adenine [A], thymine [T], guanine [G], and cytosine [C]) on a gene. The underlying principle involves labeling the four types of bases in the gene with distinct fluorescence colors. Within the gene sequencer, a laser system activates the bases in their arrayed order, where each base type yields a unique fluorescence color. Special cameras capture these fluorescent signals, which eventually elucidates the nucleotide sequence within the gene.
The advancements in science and technology have progressively refined gene sequencing techniques, moving from what we now refer to as Sanger sequencing to the more contemporary Next-Generation Sequencing (NGS) and Long-read sequencing techniques.
Service you may intersted in
1. Sanger sequencing
In 1977, the arrivals of the dideoxy chain termination method proposed by Sanger and the chemical degradation method proposed by Gilbert heralded the birth of first-generation sequencing technology. Among them, Sanger's dideoxy chain termination method (Sanger Sequencing) is the most classic and one of the most widely applied methods for detecting gene mutation in Sanger sequencing technology. Meanwhile, the chemical degradation method has gradually been phased out. Sanger sequencing, targeting the specific area of the template DNA through specific primers and incorporating dideoxy nucleotides (ddNTP) into the PCR reaction, separates extension products by electrophoresis and detects fluorescent markers, thus obtaining the base information at each position and consequently identifying specific mutations in the DNA sequence (Fig. 6). Regarded as the "gold standard" of sequencing, Sanger sequencing boasts higher sequencing accuracy than NGS and TGS. Moreover, it offers refined accuracy for 700-1000 bp read lengths and can adeptly handle repetitive sequences. However, its principal limitations are its ability to only examine one template at a time, and it is comparably slower and more time-consuming (Zhang et al., 2021).
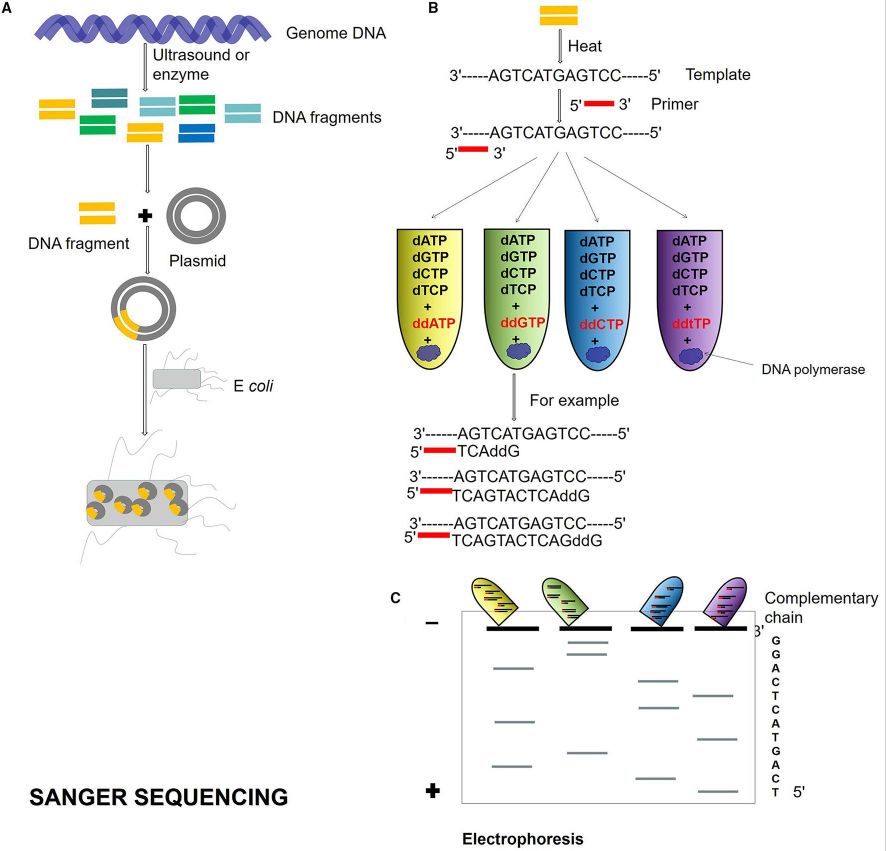 Figure.6 Sanger sequencing process (Zhang et al., 2021)
Figure.6 Sanger sequencing process (Zhang et al., 2021)
2. NGS
Next-Generation Sequencing is established on PCR technology and gene chip foundation and uses high-throughput parallel sequencing technology. It permits the simultaneous synthesis of millions of sequenced template complementary chains and accumulates sequence data. NGS primarily employs sequencing-by-synthesis (SBS) and sequencing-by-ligation (SBL) technologies. Currently, Illumina sequencing systems have become the mainstream NGS product. NGS ascertains the DNA sequence by capturing the special tag carried by the newly added bases during the DNA replication process, which is often a fluorescent molecular marker. The overall process can be divided into four main steps: library preparation, DNA cluster generation, sequencing-by-synthesis, and data analysis. Two important features of NGS are high-throughput and short read length. Its efficiency and low per base sequencing costs provide immeasurable advantages for clinical applications.
3. Long-read Sequencing
Long-read sequencing represents a milestone in the sequencing technology domain. This emerging technology, built upon Single Molecule Sequencing (SMS) and massive parallel sequencing technology, omits the need for DNA molecule PCR amplification during sequencing. It is founded on the examination of individual electrical or chemical reaction signals based on a single molecule, thus enabling the separate sequencing of each DNA molecule. While PacBio technology and Nanopore technology are included in Long-read Sequencing, one example of this technology, the Nanopore system, fastens nanopore proteins onto a resistive membrane. When nucleic acids pass through the nanopore, there are changes in electrical charge. As the diameter of the nanopores are extremely small, only allowing a single nucleic acid polymer to pass through at a time, different bases interfere with the current in distinctive ways when passing through the protein nanopore. Real-time monitoring and decoding of these current signals then determine base sequences. Long-read Sequencing, given its longer sequencing read length, can sequence original DNA samples directly, detect RNA and methylated DNA sequences directly, and has the advantage of rapid operation.
Service you may intersted in
Hybridization
1. FISH
In 1986, researchers began to utilize fluorescein isothiocyanate to label probes, which were subsequently analyzed under a fluorescence microscope, establishing the Fluorescence In Situ Hybridization (FISH) technique. In this process, exogenous nucleic acids (commonly referred to as molecular probes) pair with target DNA or RNA on cells or tissues following the principle of base complementarity, forming nucleic acid hybrid molecules. The probes are typically labeled with a report molecule, such as biotin or digoxin, on a specific nucleotide. This label instigates a chemical reaction with fluorescein-labeled specific affinity substances, enabling the capture of probe signals under a fluorescence microscope. Consequently, it becomes feasible to determine whether the target samples have undergone genetic mutations.
2. Gene microassay
The Gene Microarray technology, having experienced rapid development since the mid-1990s, stands as an advanced molecular biology technique at the crossroads of interdisciplinary synthesis. Primarily applied in disease diagnosis and treatment as well as pharmaceutical research, this innovative science has become a pivotal force in contemporary biomedical research. The fundamental principle underlying this technology involves nucleic acid sequence determination through the hybridization of fluorescently labeled nucleic acid sequences, with a set of known sequences acting as probes fixed on a solid substrate. Specifically, when a solution contains a fluorescently labeled nucleic acid sequence, such as TATGCAATCTAG, and it undergoes complementary matching with the corresponding nucleic acid probe immobilized at a specific position on the gene microarray, the probe exhibiting the strongest fluorescence intensity provides information about the sequence complementarity.
Service you may intersted in
References:
- Athanasopoulou K, Boti M A, Adamopoulos P G, et al. Third-generation sequencing: the spearhead towards the radical transformation of modern genomics. Life, 2021, 12(1): 30.
- Boonbanjong P, Treerattrakoon K, Waiwinya W, et al. Isothermal amplification technology for disease diagnosis. Biosensors, 2022, 12(9): 677.
- Cao Y, Yu M, Dong G, et al. Digital PCR as an emerging tool for monitoring of microbial biodegradation. Molecules, 2020, 25(3): 706.
- Nguyen-Dumont T, Jordheim L P, Michelon J, et al. Detecting differential allelic expression using high-resolution melting curve analysis: application to the breast cancer susceptibility gene CHEK2. BMC Medical Genomics, 2011, 4: 1-10.
- Peng B, Wang Q, Luo Y, et al. A novel and quick PCR-based method to genotype mice with a leptin receptor mutation (db/db mice). Acta Pharmacologica Sinica, 2018, 39(1): 117-123.
- Ye S, Dhillon S, Ke X, et al. An efficient procedure for genotyping single nucleotide polymorphisms. Nucleic acids research, 2001, 29(17): e88-e88.
- Zhang L, Chen F X, Zeng Z, et al. Advances in metagenomics and its application in environmental microorganisms. Frontiers in microbiology, 2021, 12: 766364.


