
 Sample Submission Guidelines
Sample Submission Guidelines
Overview of Copy Number Variations (CNVs)
What is Copy Number Variation (CNV)?
Copy Number Variations (CNVs) encompass alterations in the number of specific DNA segments within individual genomes. These variations constitute a type of genomic structural variation, typically involving segments exceeding 1 kilobase in length. These structural distinctions can arise through processes like duplication, deletion, or other modifications and frequently affect one or more genes.
CNVs represent a class of genomic structural variants, which are further categorized into two levels: microscopic and submicroscopic. Microscopic genomic structural variation primarily pertains to visible chromosomal anomalies when observed under a microscope, encompassing conditions like aneuploidy or aneuploidy, deletions, insertions, inversions, translocations, and various other structural alterations. Submicroscopic genomic structural variation, on the other hand, refers to variations in DNA fragment lengths ranging from 1 kilobase to 3 megabases, encompassing deletions, insertions, duplications, rearrangements, and inversions. Collectively, these submicroscopic changes are referred to as CNVs or CNPs (Copy Number Polymorphisms, CNP).
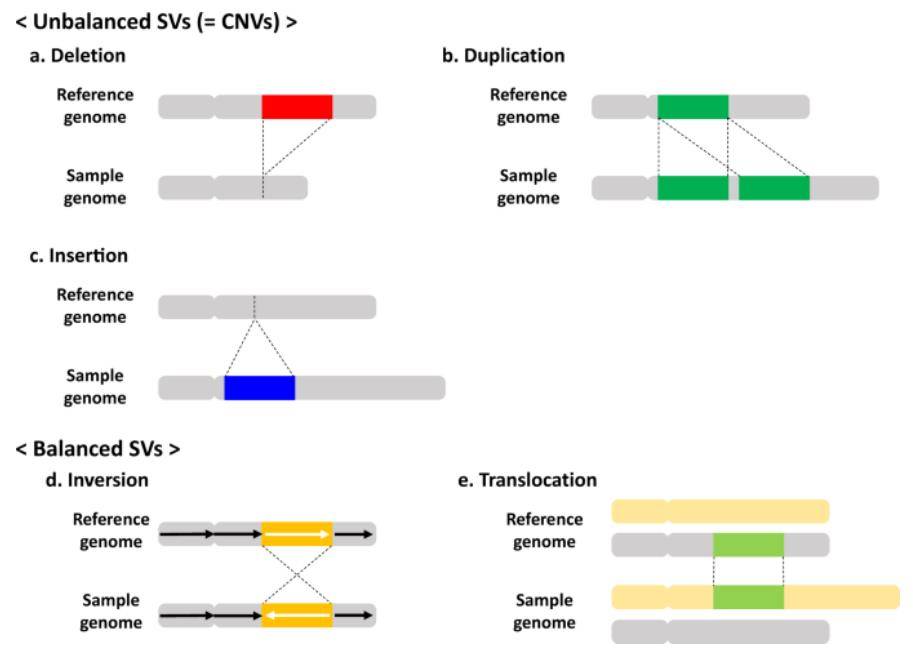 Germline copy-number variations. (Nakatochi et al., 2021)
Germline copy-number variations. (Nakatochi et al., 2021)
Mechanisms Underlying the Formation of Copy Number Variations (CNVs)
Copy Number Variations (CNVs) are genetic phenomena arising from a diverse array of genetic events. These mechanisms encompass several distinct processes. One such mechanism is replication error, were errors during DNA replication result in insertions or deletions, ultimately leading to alterations in the copy number of specific DNA segments. Another mechanism involves non-homologous recombination, characterized by the exchange of DNA segments between different chromosomes, resulting in changes in copy numbers for both involved chromosomes.
Recombination events primarily occur within specific regions of repetitive sequences known as Low Copy Repeats (LCRs). LCRs harbor various genetic elements, including genes, pseudogenes, gene fragments, retroviral sequences, and gene regulatory regions. Typically, LCRs are situated at the termini of chromosomes and along the chromosomal filaments. The size, relative orientation of LCRs, the spacing between copies, and the degree of sequence homology all influence the formation of CNVs.
However, the precise mechanisms governing CNV formation remain incompletely understood. Several mechanisms have been proposed:
- Non-Allelic Homologous Recombination (NAHR): NAHR primarily occurs in regions prone to frequent recombination. Key characteristics of NAHR regions include fragment sizes exceeding 10 kb, sequence homology surpassing 97%, well-defined sequence orientation, and LCRs located within the same chromosome.
- Non-Homologous End Joining (NHEJ): Some simpler CNVs can arise through the NHEJ mechanism, which does not necessitate precise homology between DNA breaks. Instead, NHEJ can join disparate DNA breaks, potentially leading to chromosomal rearrangements like translocations and other mutations.
- Fork Stalling and Template Switching (FoSTeS/MMBIR): FoSTeS represents a complex mechanism responsible for generating CNVs with intricate structures. This mechanism not only produces CNVs spanning several megabases but also induces rearrangements at both the gene and exon levels. These rearrangements encompass gene duplications and exon shuffling, contributing significantly to genome evolution.
The interplay of these mechanisms and their specific consequences in various contexts warrants further investigation. A comprehensive understanding of CNV formation and their implications for genetic diversity and disease etiology necessitates continued research.
CNVs and Disease
Currently, Copy Number Variations (CNVs) in the realm of disease can be broadly classified into three primary categories:
- Germ Cell-Inherited CNVs: These CNVs are inherent in the parental genome and are passed on to their offspring through germ cells. Detecting these CNVs involves examining the parents' genomes and is a crucial aspect of genetic counseling. Germ cell-inherited CNVs are linked to the development of numerous genetic disorders.
- De Novo Acquired CNVs: Unlike germ cell-inherited CNVs, these variants are not present in the parental genomes but emerge in the genomes of newborns. They can result from sudden mutational events or mutations occurring during early embryonic development. De novo acquired CNVs are also associated with the onset of various diseases.
- Somatic Cell-Produced CNVs: These CNVs arise after the embryonic single-cell phase and can stem from environmental influences, cell division, or other biological processes. Somatic CNVs can differ between various tissues within an individual and may influence the functionality and attributes of somatic cells.
Impact of Copy Number Variations (CNVs) on Gene Activity
Copy Number Variations (CNVs) dispersed across various loci in the genome can give rise to a diversity of genomic and molecular phenotypic variations, ultimately contributing to the emergence of intricate diseases, including cancer. The underlying mechanisms through which CNVs influence gene expression and consequently trigger tumorigenesis encompass the following:
- Gene Knockouts and Disruptions: The most straightforward manner in which CNVs influence gene expression is by either eliminating an entire gene or disrupting specific portions of it. This can occur through the insertion of partial sequences that encode either nonfunctional or functionally altered proteins.
- Regulatory Region Interference: CNVs can also exert their influence by interfering with the regulatory regions of a gene. Fragmentary deletions within these regulatory regions may enhance gene transcriptional activity, while fragment duplications involving promoters and adjacent areas can result in unstable rearrangements, ultimately diminishing gene expression activity. A notable example of this is the CNV-related modulation of the UGT2B17 gene in testicular cancer, which not only impacts its own activity but also influences neighboring genes.
- Dose-Dependent Effects: CNVs have the potential to directly modulate the expression of specific genes through their dosage effects. Tiny deletions or duplications of genes within CNV regions can disrupt their normal functioning. In essence, an increase in CNV copies amplifies gene expression within the affected region or nearby regions, whereas a reduction in copies diminishes gene expression.
- Structural Gene Alterations: CNVs can induce structural changes in genes, thereby affecting the products of gene expression. For instance, a prevalent CNV on chromosome 1q21.1, which is also implicated in neuroblastoma susceptibility, results in modified expression of a recently identified neuroblastoma breakpoint family (NBPF) gene transcript, NBPF23.
Methods for Detecting Copy Number Variations (CNVs)
Array-Based Comparative Genomic Hybridization (aCGH)
Array-Based Comparative Genomic Hybridization, commonly referred to as aCGH, is a powerful technique used to identify copy number variations within a genome. By co-hybridizing samples labeled with distinct fluorescent markers on a single microarray chip, aCGH allows for the visualization of deletions or amplifications in genomic DNA, both in the context of tumors and hereditary diseases across entire chromosome groups. This method utilizes specialized instruments and chips, boasting high resolution and a high degree of automation. Importantly, aCGH enables the comprehensive detection of CNVs across the entire genome in a single experiment.
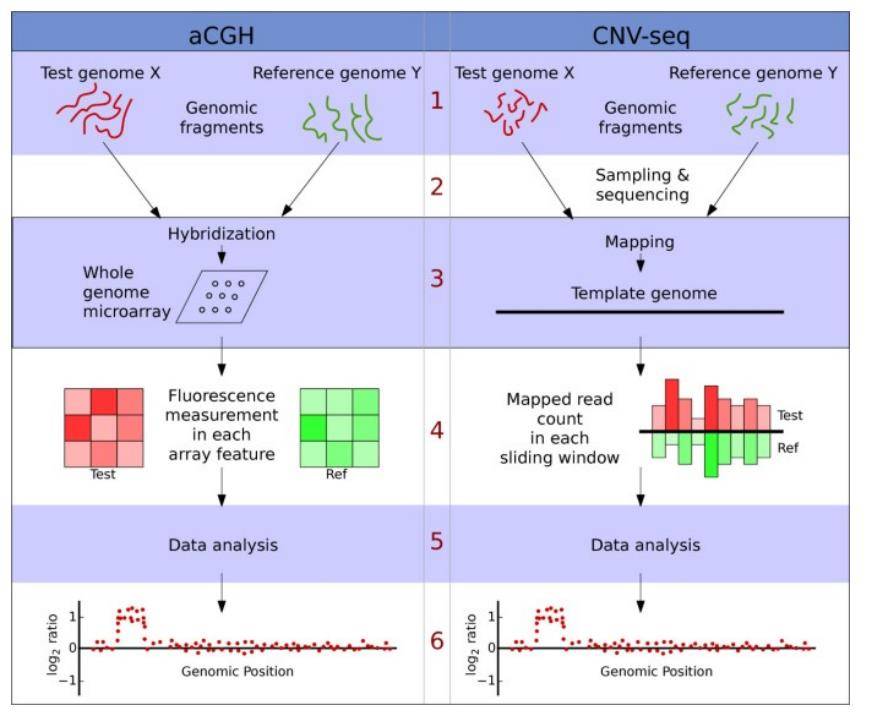 A comparison of the conceptual steps in aCGH and CNV-seq methods. (Xie et al., 2009)
A comparison of the conceptual steps in aCGH and CNV-seq methods. (Xie et al., 2009)
Single Nucleotide Polymorphism Array (SNP Array)
The Single Nucleotide Polymorphism Array, or SNP Array, employs a unique approach distinct from aCGH. Instead of a two-hybridization strategy, SNP Array involves a single hybridization event between the samples under investigation and microarray probes. It then determines the copy number at each genomic locus by analyzing the signal intensities from various samples. SNP Array offers exceptional resolution and is capable of detecting a wide range of microdeletions and microduplications, including phenomena such as uniparental disomy (UPD), heterozygous deletion (LOH), and chimerism, in addition to CNVs.
Sequencing-Based Methods
Sequencing-based techniques provide an alternative approach to CNV detection, typically using whole-genome sequencing. Similar to aCGH, these methods involve sequencing equal amounts of DNA from the sample of interest and a normal control DNA, which are then compared to a reference sequence. The copy number at each genomic locus is determined by comparing the read counts within sliding windows between the two samples. This approach enables the detection of large segments of CNVs across the entire genome.
References:
- Nakatochi, Masahiro, Itaru Kushima, and Norio Ozaki. "Implications of germline copy-number variations in psychiatric disorders: review of large-scale genetic studies." Journal of Human Genetics 66.1 (2021): 25-37.
- Xie, Chao, and Martti T. Tammi. "CNV-seq, a new method to detect copy number variation using high-throughput sequencing." BMC bioinformatics 10 (2009): 1-9.


