
 Sample Submission Guidelines
Sample Submission Guidelines
Combination of ChIP-seq and ATAC-seq Illuminates the Regulatory Landscape of the Genome
While ChIP-seq and ATAC-seq individually provide valuable insights into different aspects of chromatin biology, their combination unlocks a deeper understanding of gene regulation. Integrating ChIP-seq data with ATAC-seq data enables researchers to explore the connection between transcription factor binding and chromatin accessibility.
By comparing ChIP-seq and ATAC-seq profiles, researchers can determine whether transcription factors are bound to regions of accessible chromatin, suggesting active regulatory sites. Conversely, if transcription factor binding is detected in regions with closed chromatin, it may indicate the potential for gene regulation upon chromatin remodeling.
The combination of these techniques also aids in identifying cell-type-specific regulatory elements. The joint analysis allows researchers to distinguish between general regulatory elements found in most cell types and those that are unique to specific cell lineages, providing critical insights into cell-type-specific gene regulation and potential therapeutic targets.
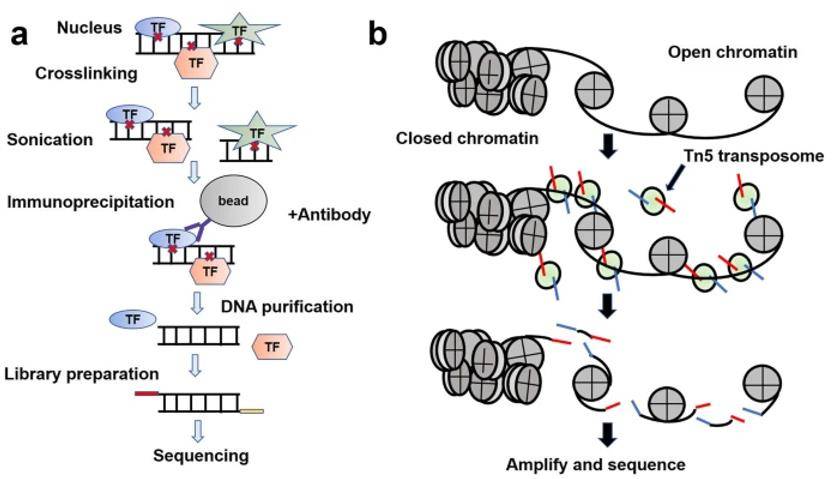 Workflows of ChIP-seq and ATAC-seq. (Ma et al., 2020)
Workflows of ChIP-seq and ATAC-seq. (Ma et al., 2020)
ChIP-seq and ATAC-seq Advance Embryo Chromatin Research
During mammalian embryonic development, various epigenetic changes occur across the genome, including DNA methylation, histone modifications, open chromatin regions, and changes in chromatin conformation. These genomic regulatory elements, such as promoters, enhancers, insulators, and motif control regions, play critical roles in directing embryonic development through interactions with cell type-specific transcription factors. Furthermore, the long-term interactions between these regulatory elements have piqued researchers' interest.
Recent innovations in ChIP-seq have enabled the study of histone modifications in very low cell numbers, such as embryos. In early mouse embryos, the histone modification H3K4me3 undergoes extensive reprogramming events. It disappears in the syncytium and reappears during the progeny's syncytial genome activation (ZGA). By optimizing ChIP-seq in embryos, researchers can delve into the intricate process of transmitting mammalian histone modifications from parents to offspring. This includes identifying differences in parental modification patterns before and after fertilization, shedding light on crucial developmental events.
Similarly, the enhanced ATAC-seq method finds application in embryonic tissues, aiding in the localization of genome-wide chromatin accessibility during critical periods of embryonic development. For instance, in preimplantation embryos, ATAC-seq has been instrumental in uncovering high-resolution chromatin changes during ZGA and the temporal dynamics of secondary syntenic genome activation (minorZGA). These epigenomic studies have also revealed unique chromatin states at different stages of embryonic development, offering valuable insights for further research into human embryonic development and potential clinical implications.
While these advances have unveiled essential aspects of early embryonic development, there are still questions that remain unanswered. Researchers seek to identify key factors responsible for regulating shifts in chromatin states and explore the role of transposons in this intricate process.
Unraveling Organ Development with ChIP-seq and ATAC-seq
Epigenome sequencing technologies have become indispensable tools in studying developmental trajectories and understanding cell fate determination. Leveraging ChIP-seq and ATAC-seq at the single-cell level enables comprehensive exploration of tissue and organ developmental dynamics. Notably, in 2018, scATAC-seq was employed to analyze clustered cells at different stages of mouse forebrain development, revealing key regulators inferred from open chromatin.
By integrating ChIP-seq, ATAC-seq, and DNase-seq with transcriptomic data in organoid models, researchers gain valuable insights into the developmental dynamics of specific cells, identify essential transcriptional regulators, and pinpoint cell clusters prone to disease susceptibility. This multi-omics approach, combining single-cell transcriptome and ATAC-seq data in organ development, lays a robust foundation for clinical disease treatment. For instance, in-depth understanding of key time points and gene regulatory networks in human hippocampal development has provided crucial information about cell populations linked to the pathology of Parkinson's, Alzheimer's, and Huntington's diseases.
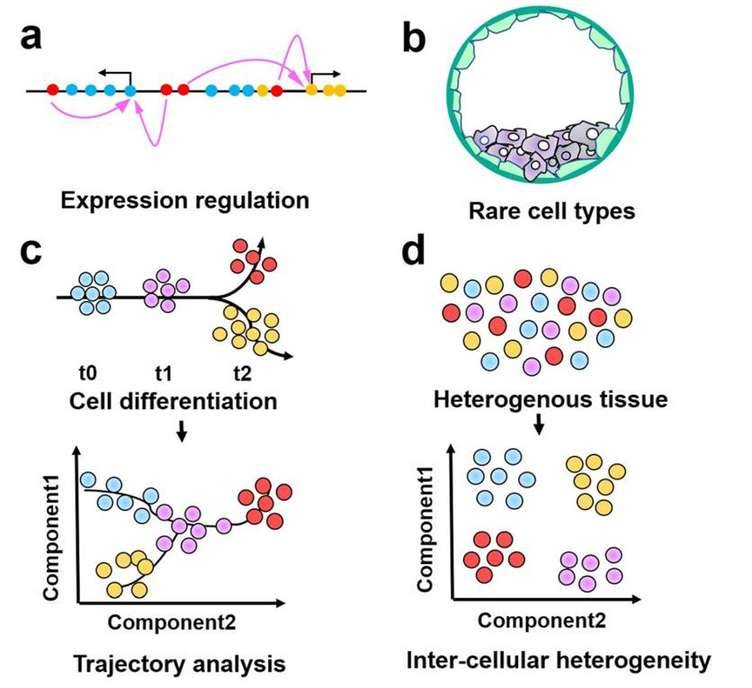 Future applications of single-cell epigenomics. (Ma et al., 2020)
Future applications of single-cell epigenomics. (Ma et al., 2020)
Beyond neurobiological investigations, chromatin dynamics analyses have also illuminated muscle development, mammary gland development, and cardiac precursor cell fate. Fine-grained studies of single-cell chromatin dynamics are paving the way for comprehensive models of human organ development, enabling researchers to trace the embryonic origin of each tissue and organ effectively.
Unveiling the Complexity of Cancer through ChIP-seq and ATAC-seq
Our current understanding of highly heterogeneous tumor tissues remains limited, encompassing variations in the tumor microenvironment, disparities between primitive primary cancers and metastases, and the evolution of tumor subclones. Crucially, immune cells in the tumor microenvironment play a significant role in immune escape and cancer cell infiltration. The emergence of single-cell ATAC-seq (scATAC-seq) and its applications offer a promising avenue to unravel the epigenetic heterogeneity driving tumor progression and identify potential therapeutic targets.
By applying scATAC-seq, researchers have identified regulatory networks governing malignant stroma and immune cells in the tumor microenvironment. In-depth exploration of individual immune cell development kinetics has enabled comparisons of T cell depletion within the tumor microenvironment before and after immunotherapy. As a result, key regulatory T-cell populations responsive to immunotherapy have been identified, facilitating personalized treatment strategies.
Integrating ChIP-seq, ATAC-seq, and DNA mutation profiles within the same cells empowers scientists to uncover novel cancer cell subclones for tailored clinical trials. Thus, comprehending chromatin regulation patterns at the single-cell level holds the potential to drive significant biomedical advancements in cancer treatment.
Furthermore, the expanded technology of ATAC-seq, including ATAC-see, offers fresh insights into tumor heterogeneity. ATAC-see enables in situ imaging of open chromatin by fluorescently labeling open motifs, providing physical evidence of the co-localization of extrachromosomal DNA (ecDNA) and ATAC-see signals. Supported by ATAC-seq and MNase-seq data, this discovery highlights the high accessibility of ecDNA, explaining the abundant expression of oncogenes located on ecDNA.
Overall, leveraging the adaptability of ChIP-seq and ATAC-seq technologies holds great promise in paving the way for targeted therapies and providing physical evidence to visualize cancer heterogeneity. These advances are set to foster comprehensive and reliable scientific discoveries in the battle against cancer.
Reference:
-
Ma, Shaoqian, and Yongyou Zhang. "Profiling chromatin regulatory landscape: insights into the development of ChIP-seq and ATAC-seq." Molecular biomedicine 1 (2020): 1-13.


