
 Sample Submission Guidelines
Sample Submission Guidelines
What is CITE-Seq?
CITE-seq (cellular indexing of transcriptomes and epitopes) represents a breakthrough in single-cell sequencing, enabling simultaneous capture of transcripts and a portion of membrane surface proteins. This innovative method supplements traditional 10X Genomics single-cell RNA sequencing by incorporating detection for cell surface proteins. Statistical analyses reveal a strong correlation between RNA- and ADT-based cell categorization, indicating minimal signal loss in ADT detection, thus facilitating accurate cell class identification. Particularly advantageous for analyses involving a high cell count prone to ambiguity, CITE-seq enhances the ability to distinguish between cell types without compromising signal integrity.
Simultaneously, cell hashing tagging technology enables the realization of multi-sample mixing for CITE-seq detection, followed by the splitting of single-cell data. This facilitates the subsequent identification and characterization of cell lines and antibody-captured surface proteins, leveraging HTO or ADT tag sequences, respectively.
Advantages of CITE-Seq
- Versatility in Protein Detection: CITE-seq boasts the capability to detect over 100 surface proteins concurrently, seamlessly integrating with commercial platforms. This surpasses the limitations of flow sorting, offering an unlimited range of antibodies.
- Enhanced Cell Typing: CITE-seq enables clearer identification of cell types, facilitates the annotation of rare cell types, and fosters the discovery of novel cell subpopulations.
- Dropout Mitigation: Integration with cell Hashing labeling technology effectively addresses the dropout issue, enhancing data quality.
- mRNA and Protein Integration: CITE-seq achieves multi-omics joint detection within the same cell, providing simultaneous mRNA and cell surface protein information. This comprehensive approach surpasses traditional single-cell RNA sequencing in informativeness.
Moreover, 10X Genomics provides a well-established ADTs-coupled labeling system and a detailed procedural framework for CITE-seq. Leveraging Feature Barcode technology, it efficiently conducts transcriptome and surface protein studies in immune cells. Additionally, compatible products such as oligonucleotide-coupled antibodies and oligonucleotide-coupled MHC further enable single-cell multi-omics analysis.
Applications of CITE-Seq
CITE-seq serves as a powerful tool for integrating protein information with genes, aiding in the precise characterization of cell states.
The Weighted Nearest Neighbor (WNN) analysis method represents a sophisticated approach for harmonizing multi-omics data within individual cells, thus yielding a comprehensive definition of cell state. This method operates on an unsupervised basis, initially computing cell-specific histological weights through cross-prediction across multiple omics layers. These weights, which encapsulate the relative significance of each histological information component, subsequently inform the distance metric between cells, resulting in the construction of a WNN neighborhood graph—termed the WNN graph—depicting the multi-omics landscape within single cells. The WNN graph finds utility across various downstream analysis tasks in single-cell data analysis, including UMAP visualization, clustering analysis, and the construction of cytomimetic time series.
- Cell Identity Determination
Initially, RNA data were individually processed, and RNA+ADT data were jointly analyzed using the Weighted Nearest Neighbor (WNN) approach. Annotation utilizing classical markers revealed that the incorporation of ADT data during analysis led to enhanced cell type differentiation and annotation of a broader range of cell types. Notably, the introduction of ADT down-conversion highlighted more distinct differences among T cell subpopulations compared to RNA alone. Combining RNA and ADT weight violin plots within each cell type indicated elevated ADT weights within Macrophages (MPs) and T cell subpopulations. Integrating ADT for dimensionality reduction demonstrated improved discrimination between subpopulations compared to RNA alone, offering extensive protein validation and identification of critical surface markers for each subpopulation. Prior studies have corroborated that CITE-seq enhances population resolution, effectively distinguishing similar subpopulations and even unveiling novel PD-L1/PD-L2+ macrophage populations correlated with clinical outcomes.
- Subpopulation Delineation
Subsequently, subpopulation delineation and validation of scRNA-seq findings were conducted. Given the higher protein weights observed in T cells, further subdivision of T cell subpopulations was pursued. Validation of scRNA-identified subpopulations via CITE-seq results revealed a more pronounced separation of subpopulations in UMAP plots based on protein data. RNA and ADT weight violin plots highlighted the role of protein expression in distinguishing subpopulations during T cell subdivision. Additionally, extensive protein validation facilitated the identification of key surface markers for each subpopulation.
- Mechanism Exploration through Functional Analysis
Further subdivision of NK subgroups revealed that the incorporation of ADT features facilitated better subgroup discrimination, with distinct protein marker expression observed among subgroups. For instance, cluster1 exhibited specific expression of the CD8a protein marker, while cluster5 showed significant enrichment in the NK cell-mediated toxicity pathway. Notably, there were no significant differences in CD8A gene expression among clusters.
Case Study: Liver Single-Cell Analysis
In a groundbreaking endeavor, the research team merged single-cell CITE-seq with complementary technologies to chart the spatial proteome of both human and mouse liver single cells, shedding light on the lipid-associated macrophage population. Delving into mRNA and protein expression at single-cell granularity, 18 samples underwent CITE-seq and subsequent data analysis, culminating in the effective identification of 17 distinct cell types. Concurrently, CITE-seq's ADT antibody recognition system adeptly captured Kupffer cells, and through meticulous cell population analysis, the VSIG4 protein emerged as the premier marker for human Kupffer cells within the CITE-seq dataset.
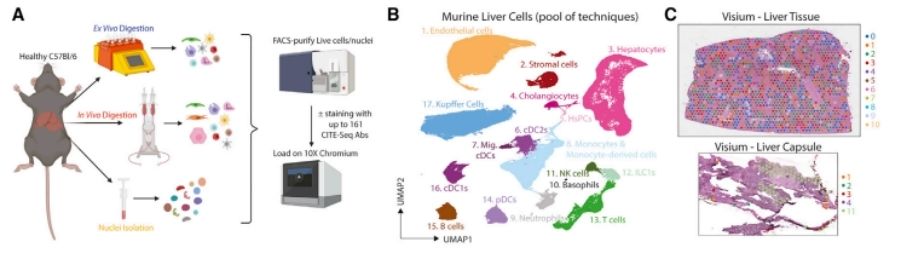 A proteogenomic atlas of the healthy murine liver. (Guilliams et al., 2022)
A proteogenomic atlas of the healthy murine liver. (Guilliams et al., 2022)
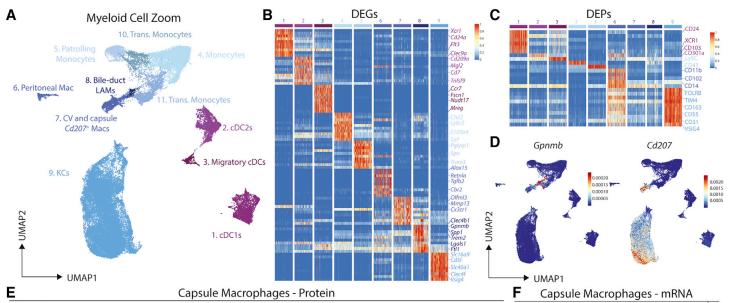 A population of macrophages reside around the bile duct in the healthy murine liver. (Guilliams et al., 2022)
A population of macrophages reside around the bile duct in the healthy murine liver. (Guilliams et al., 2022)
Case Study: Single-Cell Investigation of Lung Cancer
This study presents a comprehensive analysis of the largest non-small cell lung cancer (NSCLC) cohort to date, employing both single-cell RNA sequencing (scRNA-seq) and CITE-seq methodologies. By delving into immune responses shared among patients, the research identified common factors while uncovering distinct immune modulatory effects of tumor mutation load and TP53 mutations. Consequently, refined models were crafted to predict immunotherapy responses with greater accuracy.
The tumor microenvironment's transcriptional landscape was meticulously explored through scRNA-seq and CITE-seq analyses, revealing a spectrum of transcriptional states.
Specifically, CITE-seq data highlighted unique characteristics of CD8+ T cells, resembling natural killer (NK) cells, thereby delineating T cell subsets within both normal and cancerous tissues. Notably, cancer tissues exhibited a broader array of T cell subsets, encompassing activating, cycling, and regulatory T cells.
Leveraging insights from CITE-seq data and other single-cell multi-omics investigations, a single-cell immune response map was constructed for lung cancer. This map delineated an immune activation signature, pinpointing the interplay between tumor-associated antigenic loads and driver mutation states. Furthermore, a model was developed utilizing the LCAM axis, offering a more direct measure of antigen specificity and anti-tumor immune activation. This refined model promises enhanced precision in defining immune activation within antigen-specific tumors.
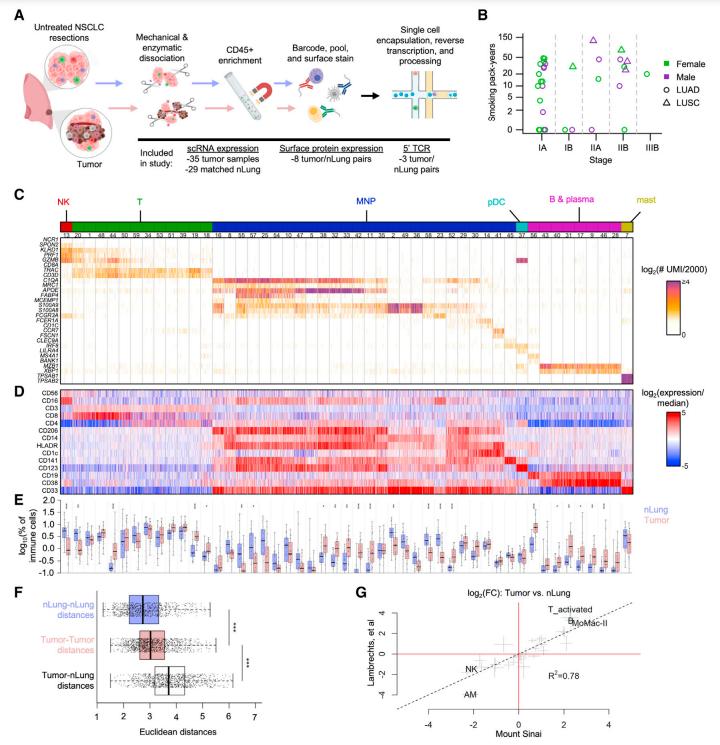 scRNA-seq and CITE-seq establish the diversity of transcriptional states in the tumor microenvironment. (Leader et al., 2021)
scRNA-seq and CITE-seq establish the diversity of transcriptional states in the tumor microenvironment. (Leader et al., 2021)
References:
- Guilliams, Martin, et al. "Spatial proteogenomics reveals distinct and evolutionarily conserved hepatic macrophage niches." Cell 185.2 (2022): 379-396.
- Leader, Andrew M., et al. "Single-cell analysis of human non-small cell lung cancer lesions refines tumor classification and patient stratification." Cancer cell 39.12 (2021): 1594-1609.


