
 Sample Submission Guidelines
Sample Submission Guidelines
A Guide to Cancer Whole Exome Sequencing
Whole Exome Sequencing (WES) has emerged as a transformative force, enabling the comprehensive analysis of the coding regions of genes linked to diseases and population evolution. When coupled with the vast exome data available in public databases, WES becomes a potent tool to unravel the intricate relationships between genetic variants, diseases, and the underlying mechanisms. While its applications are diverse, the application of WES in cancer research shines the brightest due to its complexity and the unprecedented opportunities it offers to tackle one of humanity's most challenging foes.
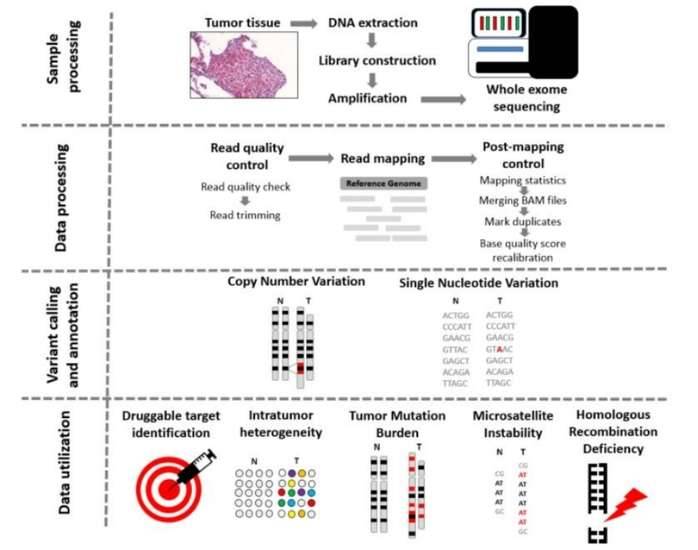 From tissue to data—steps of whole exome sequencing. (Bartha et al., 2020)
From tissue to data—steps of whole exome sequencing. (Bartha et al., 2020)
Sample Requirements and DNA Extraction: Foundations of Accurate Analysis
Conducting Cancer Whole Exome Sequencing (CWES) necessitates meticulous sample selection and DNA extraction to ensure the fidelity and relevance of results. Tumor tissues, formalin-fixed paraffin-embedded (FFPE) samples, and peripheral blood are the common sample types used. However, a pivotal aspect of CWES is the pairing of tumor samples with matched normal samples. This pairing not only mitigates the impact of genetic background but also enables the identification of somatic mutations unique to the tumor.
The process of DNA extraction is a delicate art aimed at preserving DNA integrity while eliminating contaminants. It involves a series of steps such as cell lysis, enzymatic treatment, nucleic acid separation, and purification. Notably, the challenges posed by FFPE samples demand special attention. Formaldehyde-induced cross-linking, DNA adduct formation, and modifications leading to false positives underscore the need for a strategic approach in handling such samples.
Mastering Library Construction: Technical Nuances
Library construction in CWES is a multidimensional endeavor where precision is paramount. Several technical aspects must be meticulously addressed to guarantee accurate and dependable sequencing outcomes.
- Optimizing Fragment Transformation Efficiency
Library construction's success hinges on achieving high fragment transformation efficiency—converting a maximal number of starting DNA fragments into measurable library fragments. A key determinant of success is library complexity, which translates to richer sequencing data coverage of the target regions. Achieving this requires meticulous consideration of starting DNA quantity, quality, and fragment conversion efficiency. Rigorous testing and evaluation are indispensable to realize optimal fragment transformation efficiency. - Taming Background Noise
In the intricate dance of Next-Generation Sequencing (NGS), errors creep in, and background noise muddles the signal. Background noise arises from various sources including PCR errors, sequencing inaccuracies, and complex genomic regions. Addressing this requires ingenuity—unique molecular tags attached to DNA fragments during library construction act as sentinels, helping discern real mutations from erroneous signals. This clever strategy effectively curbs background noise, elevating the accuracy of mutation detection. - Defying Contamination
The specter of sample cross-contamination haunts library construction, posing a grave threat to the veracity of results. Misplaced labels or data crossovers during analysis can skew findings. To thwart this, the employment of double-end indexing during library construction becomes imperative. Particularly relevant in platforms like Illumina, where index hopping can occur, this approach safeguards against sample cross-contamination and upholds the integrity of results. - Neutralizing Amplification Bias
Genomic regions should be equally represented in the final sequencing dataset to evade preferential amplification and skewed insights. Achieving this balance demands the selection of a high-fidelity enzyme and the careful calibration of PCR amplification cycles. By minimizing amplification bias, a more accurate portrayal of genetic variants emerges.
Data Analysis: Unraveling the Genetic Tapestry
The data analysis phase is where the true power of CWES is harnessed. It involves deciphering the complex genomic alterations that drive cancer progression and identifying potential therapeutic targets.
- Variant Prioritization
Sorting through the vast array of variants requires strategic prioritization. Variants are classified based on their functional impact, frequency in databases, and relevance to cancer biology. Pathogenic and likely pathogenic variants take precedence as potential driver mutations. - Functional Annotation and Pathway Analysis
Functional annotation tools unravel the potential consequences of identified variants. Predicting their impact on protein structure and function aids in assessing their role in carcinogenesis. Pathway analysis contextualizes these variants, highlighting altered signaling pathways crucial for cancer development. - Integration with Clinical Data
Marrying genomic insights with clinical data creates a comprehensive patient profile. Patient history, treatment response, and outcome data guide the identification of actionable mutations and potential treatment strategies. This integration lays the foundation for precision oncology, tailoring therapies to a patient's unique genetic landscape.
Reference:
- Bartha, Áron and Balázs Győrffy. "Whole Exome Sequencing Data Analysis Algorithms in Cancer Diagnostics." (2020).