
 Sample Submission Guidelines
Sample Submission Guidelines
Low-Coverage Sequencing Effectively IDs Novel Variants in Underrepresented Populations
March 26, 2021 | Christie Rizk
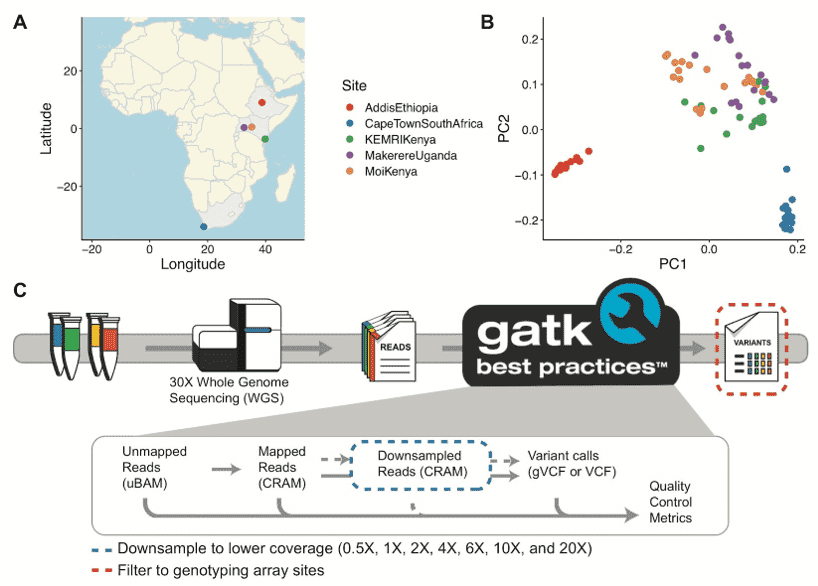
NEW YORK – An international group of researchers has shown that low-coverage sequencing can effectively identify novel variants in the genomes of individuals from populations that are currently underrepresented in genomic databases, and can help overcome challenges presented by common genotyping arrays.
In a study published on Thursday in the American Journal of Human Genetics, the researchers noted that most genetic studies use genotyping arrays and sequenced reference panels that best capture variation most common in European ancestry populations. In order to compare data generation approaches that were best suited for underrepresented populations, therefore, the researchers sequenced the whole genomes of 91 individuals as part of the Neuropsychiatric Genetics of African Population-Psychosis (NeuroGAP-Psychosis) study with participants from Ethiopia, Kenya, South Africa, and Uganda.
They sequenced these genomes to high coverage, then used a down-sampling approach to evaluate the quality of genome-wide association study (GWAS) arrays versus low-coverage sequencing, by calculating the concordance of imputed variants from these technologies with those from deep whole-genome sequencing data. In doing this, the researchers found that low-coverage sequencing at a depth of 4x or more captured variants of all frequencies more accurately than all the commonly used GWAS arrays they investigated, and at a comparable cost. Further, they noted, the lower depths of sequencing of 0.5x to 1x performed comparably to commonly used low-density GWAS arrays.
The researchers also found that low-coverage sequencing was also sensitive to novel variation: 4x sequencing detected 45 percent of singletons and 95 percent of common variants that had been previously identified in high-coverage African whole genomes.
"Our work indicates that it's high time to switch over from microarrays to low-coverage whole genome sequencing for complex disease studies. This is especially true for populations underrepresented in genetic studies for two big reasons," said first author and Broad Institute researcher Alicia Martin.
"First, we can bridge the gap between costly rare variant studies usually done in families and cheaper common variant studies to more fully understand the genetic architecture of common diseases," Martin said. "Second, unlike microarrays, low-coverage sequencing provides the opportunity to build lasting genomic resources that will facilitate future studies in populations that currently lack good reference datasets."
From their various analyses, the researchers found that 4x sequencing outperformed all the GWAS arrays they evaluated, including dense arrays such as the H3Africa array, which was designed to capture African variation. The 4x sequencing was also comparable in price to high-density arrays that assayed millions of SNPs and indels across the allele frequency spectrum.
They further found that 1x sequencing was among the more affordable options, costing less and performing similarly to or better than commonly used lower-density arrays such as the Illumina GSA. They also noted that the GSA is composed of variants that are most common in European populations and so it's therefore not the most appropriate technology for studies of participants with primarily non-European ancestry.
Aside from cost, low-coverage sequencing had several distinct advantages compared to GWAS arrays, particularly more accurate identification of genetic variation across the allele frequency spectrum in underrepresented populations. In the NeuroGAP-Psychosis data, the researchers found that 38 percent of common variants could not be imputed from the 1000 Genomes Phase III data, most likely because of a lack of eastern and southern African diversity in that panel.
Among rare variants, the fact that 4x sequencing detected nearly half of all singletons was especially appealing for disease studies. Previous work in psychiatric genetics has shown that while common variants explain most of the SNP heritability for schizophrenia, for example, exome studies are also revealing partially converging genetic signatures that are informative for severe psychiatric disorders, they said. Sequencing technologies that can bridge the gap between rare and common variants, therefore, will be critical in fully elucidating their genetic architectures by refining causal variants, detecting enriched variation, and identifying rare variants with large effects.
Post-GWAS methodological advances with low-coverage sequencing data can facilitate these analyses, the researchers noted.
"We will be using low-coverage sequencing in major studies underway, including the NeuroGAP-Psychosis study of 40,000 study participants from Ethiopia, Kenya, South Africa, and Uganda, as well as the Populations Underrepresented in Mental illness Association Studies (PUMAS) umbrella project that will include 120,000 sequenced Latin American and African populations," Martin added.
More info at: https://www.genomeweb.com/sequencing/low-coverage-sequencing-effectively-ids-novel-variants-underrepresented-populations#.YGKSBa8zY2w


