
 Sample Submission Guidelines
Sample Submission Guidelines
Genome-Wide Detection of Cytosine Methylation by Single Molecule Real-Time Sequencing
Tse OYO, Jiang P, Cheng SH, Peng W, Shang H, Wong J, Chan SL, Poon LCY, Leung TY, Chan KCA, Chiu RWK, Lo YMD
PNAS February 02, 2021 118 (5) e2019768118
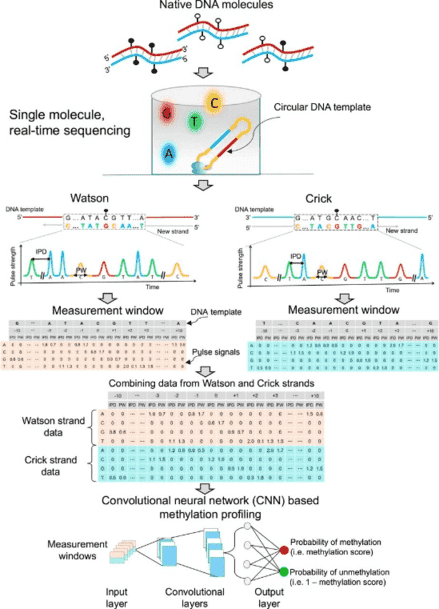
Abstract
5-Methylcytosine (5mC) is an important type of epigenetic modification. Bisulfite sequencing (BS-seq) has limitations, such as severe DNA degradation. Using single molecule real-time sequencing, we developed a methodology to directly examine 5mC. This approach holistically examined kinetic signals of a DNA polymerase (including interpulse duration and pulse width) and sequence context for every nucleotide within a measurement window, termed the holistic kinetic (HK) model. The measurement window of each analyzed double-stranded DNA molecule comprised 21 nucleotides with a cytosine in a CpG site in the center. We used amplified DNA (unmethylated) and M.SssI-treated DNA (methylated) (M.SssI being a CpG methyltransferase) to train a convolutional neural network. The area under the curve for differentiating methylation states using such samples was up to 0.97. The sensitivity and specificity for genome-wide 5mC detection at single-base resolution reached 90% and 94%, respectively. The HK model was then tested on human–mouse hybrid fragments in which each member of the hybrid had a different methylation status. The model was also tested on human genomic DNA molecules extracted from various biological samples, such as buffy coat, placental, and tumoral tissues. The overall methylation levels deduced by the HK model were well correlated with those by BS-seq (r = 0.99; P < 0.0001) and allowed the measurement of allele-specific methylation patterns in imprinted genes. Taken together, this methodology has provided a system for simultaneous genome-wide genetic and epigenetic analyses.
More info at: https://www.pnas.org/content/118/5/e2019768118


