
 Sample Submission Guidelines
Sample Submission Guidelines
Bioinformatics Workflow of Whole Exome Sequencing
Overview of WES
The advent of next-generation sequencing (NGS) has greatly accelerated genomics research, which produces millions to billions of sequence reads at a high speed. Currently, available NGS platforms include Illumina, Ion Torrent/Life Technologies, 454/Roche, Pacific Bioscience, Nanopore, and GenapSys. They can produce reads of 100-10,000 bp in length, enabling sufficient coverage of the genome at a lower cost. But faced with the enormous amount of sequence data, how do we best deal with them? And what are the most appropriate computational methods and analysis tools for this purpose? In this review, we focus on the bioinformatics pipeline of whole exome sequencing (WES).
Exome sequencing refers to a genomic analysis methodology that involves sequencing the entirety of an organism's genomic exonic regions. This is accomplished by enriching DNA in the exome region through sequence capture or target technology, followed by high-throughput sequencing. Representing just about 1% of the genome (roughly 30MB), the exomic region contains approximately 85% of pathogenic mutations.
The majority of functional variations related to an individual's phenotype are predominantly located in the chromosomal exonic region. For genetic researchers attempting to uncover the causes of over 6,800 rare diseases, exome sequencing provides a valuable tool for identifying Single Nucleotide Variants (SNVs), small insertions and deletions (InDels), as well as rare primary mutations which may elucidate complex hereditary diseases.
Bioinformatics analysis of WES plays a pivotal role in biological research, the exploration of genetic diseases, and their diagnosis and subsequent treatments. This has spurred scientific advancement and crafted new pathways to improving human health. With continual progression in technology and enhancement of analytical tools, the potential of WES bioinformatics stands to be further tapped and actualized.
Service you may intersted in
Bioinformatics Workflow of WES
You can read the article principle and workflow of WES to know more about WES. You can read the article principle and workflow of whole exome sequencing to know more about WES. The key focus of this paper is to provide a comprehensive overview of the bioinformatics analytical workflow followed post-exome sequencing. A typical workflow of WES analysis includes these steps: raw data quality control, preprocessing, sequence alignment, post-alignment processing, variant calling, variant annotation, and variant filtration and prioritization. They will be discussed below.
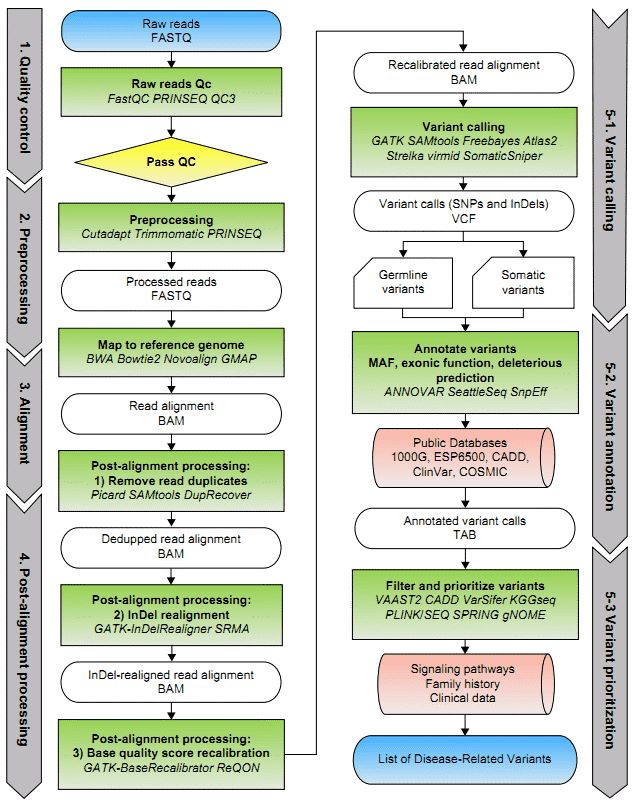 Figure 1. A general framework of WES data analysis (Bao et al. 2014).
Figure 1. A general framework of WES data analysis (Bao et al. 2014).
Raw data quality control
The generation of sequencing data involves multiple procedures such as DNA extraction, library construction, and the sequencing process itself. These procedures, however, can result in data of insufficient quality or data that is inherently invalid, necessitating a thorough quality control assessment of the raw, post-sequencing data output. The implementation of rigorous quality management paves the way for the production of high-quality sequencing data, which in turn contributes to the optimization of subsequent bioinformatics analysis procedures such as sequence alignment and variant detection. Consequently, this enhances both the efficiency and the accuracy of data analysis.
Sequence data generally have two common standard formats: FASTQ and FASTA. FASTQ files can store Phred-scaled base quality scores to better measure sequence quality. It is, therefore, widely accepted as the standard format for NGS raw data. There are multiple tools developed to assess the quality of NGS raw data, such as FastQC, FastQ Screen, FASTX-Toolkit, and NGS QC Toolkit.
Read QC parameters:
Base quality score distribution
Sequence quality score distribution
Read length distribution
GC content distribution
Sequence duplication level
PCR amplification issue
Biasing of k-mers
Over-represented sequences
Data preprocessing
With a comprehensive read QC report (generally involves the above parameters), researches can determine whether data preprocessing is necessary. Preprocessing steps generally involve 3' end adapter removal, low-quality or redundant read filtering, and undesired sequence trimming. Several tools can be used for data preprocessing, such as Cutadapt and Trimmomatic. PRINSEQ and QC3 can achieve both quality control and preprocessing.
Data preprocessing exists not only to mitigate data noise and reduce false-positive results, but also to streamline subsequent analysis processes. Preprocessing involves numerous critical steps such as quality control, technical pollutant removal, filtering of low-frequency sequences, and redundancy elimination. The ultimate aim is to convert the preprocessed data into a format amenable to subsequent analysis, trailing into formats like FASTQ and BAM. This paves the way for a more detailed bioinformatics analysis.
Sequence alignment
By performing sequence alignment, it is possible to establish the genomic location of each fragment within the exome sequencing data. This proves to be priceless in the identification of exon regions, gene structure, and functional elements. Moreover, sequence alignment plays a crucial role in various aspects such as detecting variants, facilitating gene expression analysis, and allowing data quality assessment.
Selecting an appropriate alignment tool becomes necessary and is primarily contingent upon the experimental designs and data types. Widely used tools for this purpose include Bowtie2, BWA, and STAR. For DNA sequencing data, Bowtie2 or BWA is the typical choice while STAR is frequently employed for RNA sequencing data.
There are algorithms for shot reads mapping, including Burrows-Wheeler Transformation (BWT) and Smith-Waterman (SW) algorithms. Bowtie2 and BWA are two popular short reads alignment tools that implement BWT (Burrows-Wheeler Transformation) algorithm. MOSAIK, SHRiMP2, and Novoalign are important short reads alignment tools that are implementations of SW algorithm with increased accuracy. Additionally, multithreading and MPI implementations allow significant reduction in the runtime. Of all the tools mentioned above, Bowtie2 is outstanding by fast running time, high sensitivity, and high accuracy.
Post-alignment processing
After reads mapping, the aligned reads are post-processed so as to remove undesired reads or alignment, such as reads exceeding a defined size and PCR duplicates. Tools such as Picard MarkDuplicates and SAMtools can distinguish PCR duplicates from true DNA materials. Subsequently, the second step is to improve the quality of gapped alignment via indel realignment. Some aligners (such as Novoalign) and variant callers (such as GATK HaplotypeCaller) involve indel alignment improvement. After indel realignment, BQSR (BaseRecalibrator from the GATK suite) is recommended to improve the accuracy of base quality scores prior to variant calling.
The post-alignment processing effectively identifies and filters out low-quality sequence fragments, thereby enhancing data usability and lessening the computational burden in subsequent analyses. By optimizing such post-alignment processing, the reliability and consistency of data can be maximized. This pivotal step ensures that the results of subsequent bioinformatics analyses are more credible.
Variant calling
Variant calling is a crucial process in identifying single nucleotide polymorphisms (SNPs), insertion-deletion mutations (Indels), and other genomic variations, significantly contributing to the discovery of potential pathogenic variations possibly related to diseases. Through variant calling, the genotypes of specimens can be accurately evaluated, categorizing both heterozygous and homozygous mutations. The outcomes of variant calling, therefore, serve as a foundational basis for subsequent variant annotation and filtering. As such, the precision and comprehensiveness of variant calling are pivotal for the entirety of the analytical procedure.
Specialized variant calling software, such as GATK, Samtools, and VarScan, are utilized in the post-alignment sequencing data for variant calling. These software applications discern differences between the sample and the reference genome by statistically evaluating the base information at each locus, subsequently generating a set of candidate variants.
The variant analysis is important to detect different types of genomic variants, such as SNPs, SNVs, indels, CNVs, and larger SVs, especially in cancer studies. It is vital to distinguish somatic from germline variants. Somatic variants present only in somatic cells and are tissue-specific, while germline variants are inherited mutations presented in the germ cells and are linked with patient's family history. Variant calling is used to identify SNP and short indels in exome samples. The common variant calling tools are listed in Table 1. Some studies have evaluated these variant callers. Liu et al. recommended GATK, and Bao et al. recommended a combination of Novoalign and FreeBayes.
Table 1. The common variant calling tools.
| Variant calling | Tools |
|---|---|
| Germline variant calling | GATK, SAMtools, FreeBayes, Atlas2 |
| Somatic variant detection | GATK, SAMtools mpileup, Issac variant caller, deepSNV, Strelka, MutationSeq, MutTect, QuadGT, Seurat, Shimmer, SolSNP, jointSNVMix, SomaticSniper, VarScan2, Virmid |
Variant annotation
After variants are identified, they need to be annotated for better understanding disease pathogenesis. Variant annotation generally involves information about genomic coordinates, gene position, and mutation type. Many studies focus on the non-synonymous SNVs and indels in the exome, which account for 85% of known disease-causing mutations in Mendelian disorders and a great deal of mutations in complex diseases.
Primarily, mutation annotation includes genomic coordinate transformation, mutation type annotation, functional impact prediction, gene and pathway annotation. The conversion of the genomic coordinates of a mutation onto a reference genome assures the precision and comparability of the annotation results.
The identified mutations need further annotation by type, which can include Single Nucleotide Polymorphisms (SNPs), Insertions/Deletions (Indels), Copy Number Variations (CNVs), and structural variations. Determining the genes and relevant pathways where a mutation is found involves annotating the mutation's impact on the gene, gene function classifications, regulatory elements, and more. Comparing the annotation results to public databases such as ClinVar, dbSNP, and OMIM aids in garnering known mutation information. Combined with clinical database information, one can further assess the clinical significance of a mutation.
Besides the basic annotation, there are many databases that can provide additional information about the variants. ANNOVAR is a powerful tool that combines over 4,000 public databases for variant annotation, such as dbSNP, 1000 Genomes, and NCI-60 human tumor cell line panel exome sequencing data. This tool can be used for minor allele frequency (MAF) prediction, deleterious prediction, indication of conservation of the mutated site, experimental evidence for disease variant, and prediction scores from GERP, PolyPhen, and other programs. Other common databases include OncoMD, OMIM, SNPedia, 1000 genomes, bdSNP, and personal genome variants.
Variant annotation, by associating variations with known genes, functional areas, and pathway information, assists in interpreting the functional impact of variations, such as alterations to protein structure or function. Additionally, variant annotations can sift through variations to identify candidates for pathogenic variations, thereby reducing the scope of analysis, and guiding the prioritization of potential disease-related variations. Providing a deeper biological interpretation through the annotation of function and impact of variations assists in understanding the relationship between the variant and the phenotype.
Variant filtration and prioritization
WES can generate thousands of variant candidates. The number can be reduced by variant prioritization, to generate a short but prior candidate mutation list for further experimental validation. Variant prioritization involves three steps: 1) removal of less reliable variant calls; 2) depletion of common variants (due to the assumption that rare variants are more likely to cause disease); 3) prioritization of variants relative to the disease using discovery-based and hypothesis-based approaches. The available tools for variant filtration and prioritization include VAAST2, VarSifer, KGGseq, PLINK/SEQ, SPRING, GUI tool, Gnome, and Ingenuity Variant Analysis.
Variant annotation serves the purpose of correlating identified mutations with known genes, functional regions, and pathways. By doing so, we can explore the functional impact of the variations, for instance, whether they induce alterations to protein structure or function. Variant annotation allows for the sifting of disease-causing mutations from the pool of identified variants, thereby narrowing down the scope of the analysis. This step aids in prioritizing potential disease-related mutations. Detailed examination into the function and effects of these annotated variations offers a deeper biological interpretation, thus facilitating our understanding of the link between these variations and phenotypic predispositions.
When filtering and prioritizing variants, the process often commences with a quality control phase, where detected variants possibly resulting from sequencing errors or other non-pathogenic factors are excluded. Common filtering criteria include the depth of sequencing, base quality, and heterogeneity. Subsequently, filtering is performed for common polymorphic variants.
Further filters are then applied based on the functional impact of the variants, such as non-synonymous and synonymous mutations, retaining primarily those variants likely affecting protein structure and function. Variants are then scrutinized in relation to the implicated genes, with priority given to variants occurring in genes known to be disease-associated. Once variants are sieved through these filters, they are ranked according to their probability and potential pathogenicity, taking into consideration factors such as functionality, location, frequency, and clinical implications of the variants. Ultimately, variants of high priority are selected for further validation processes, functional studies, or clinical diagnoses to establish their relevance to diseases and their biological functionality.
Variant filtering can mitigate false-positive variants introduced due to sequencing or analytical errors, thereby elevating the accuracy of variant calling. By establishing stringent filtering criteria and prioritizing strategies, potentially pathogenic variants are intentionally positioned for immediate consideration, accelerating the discovery of disease-associated variants.
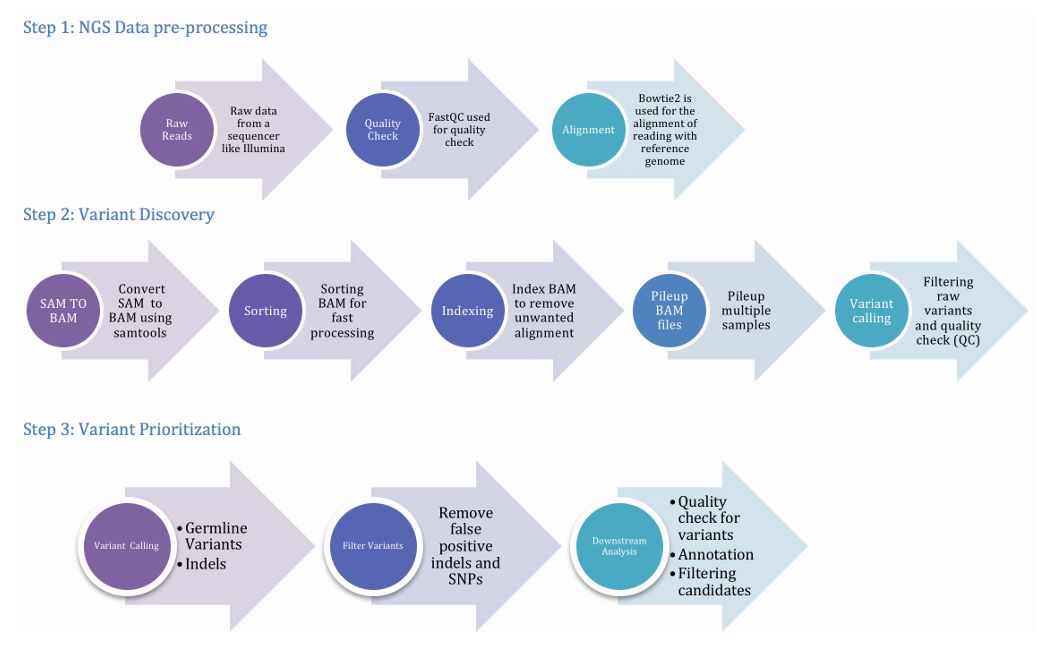 Figure 2. The pipeline involving three important phases, viz. preprocessing, variant discovery and prioritization of variants. (Meena et al., 2017)
Figure 2. The pipeline involving three important phases, viz. preprocessing, variant discovery and prioritization of variants. (Meena et al., 2017)
Data Management
In addressing data storage, the vast volume of data generated by Next Generation Sequencing (NGS) technologies may overwhelm traditional storage solutions. Consequently, the consideration of cloud storage services, such as Amazon S3, arises. These services offer nearly limitless storage capacity and operate on a pay-as-you-go model, accommodating usage fluctuations. Commercial providers like Illumina also offer cloud-based data storage services within their NGS environments, facilitating expedited access to genomic aberrations and aiding in medical diagnostics.
To economically store the extensive genomic sequencing data, compression of sequencing data becomes a viable approach. Various data compression techniques have been developed, including naive encoding, dictionary-based compression, statistical methods, and reference genome compression. For instance, the CRAM format provides an efficient compression method, significantly reducing the required storage space.
Regarding data sharing, international databases such as EBI and NCBI offer capabilities for storing and accessing large datasets. However, as data volume escalates, the sustainability of data sharing becomes a concern. Additionally, platforms like the ICGC Cancer Genome Portal and Oncomine have been established to promote data sharing. These platforms feature web-based interfaces for searching and visualizing genomic and clinical data, thereby fostering collaborative research efforts.
Conclusion
In summary, the bioinformatics analysis of whole exome sequencing is currently experiencing a period of rapid development, holding immense potential in genetic research and clinical applications. With further technological advancements and refinements in analytical methodologies, we anticipate unraveling more mysteries of genetic diseases in the future, thus enabling the provision of increasingly personalized medical interventions for patients.
If you are interested in the whole exome sequencing provided by CD Genomics, please feel free to contact us. We provide full whole exome sequencing service package, including sample standardization, exome capture, library construction, high-throughput sequencing, raw data quality control, and bioinformatics analysis. We can tailor this pipeline to your research interest.
References:
- Bao R, Huang L, Andrade J, et al. Review of current methods, applications, and data management for the bioinformatics analysis of whole exome sequencing. Cancer informatics, 2014, 13: CIN. S13779.
- Meena N, Mathur P, Medicherla K M, et al. A Bioinformatics Pipeline for Whole Exome Sequencing: Overview of the Processing and Steps from Raw Data to Downstream Analysis. bioRxiv, 2017: 201145.
- Xu H, DiCarlo J, Satya RV, Peng Q, Wang Y. Comparison of somatic mutation calling methods in amplicon and whole exome sequence data. BMC Genomics. 2014, 15:244.
- Lelieveld S H, Veltman J A, Gilissen C. Novel bioinformatic developments for exome sequencing. Human genetics, 2016, 135: 603-614.


