
 Sample Submission Guidelines
Sample Submission Guidelines
Bioinformatics Analysis of Small RNA Sequencing
Small RNAs are important functional molecules in organisms, which have three main categories: microRNA (miRNA), small interfering RNA (siRNA), and piwi-interacting RNA (piRNA). They are less than 200 nt in length and are often not translated into proteins. Small RNA generally accomplishes RNA interference (RNAi) by forming the core of RNA-protein complex (RNA-induced silencing complex, RISC). Small RNA sequencing, an example of targeted sequencing, is a powerful method for small RNA species profiling and functional genomic analysis. Here, we present the guidelines for bioinformatics analysis of small RNA sequencing.
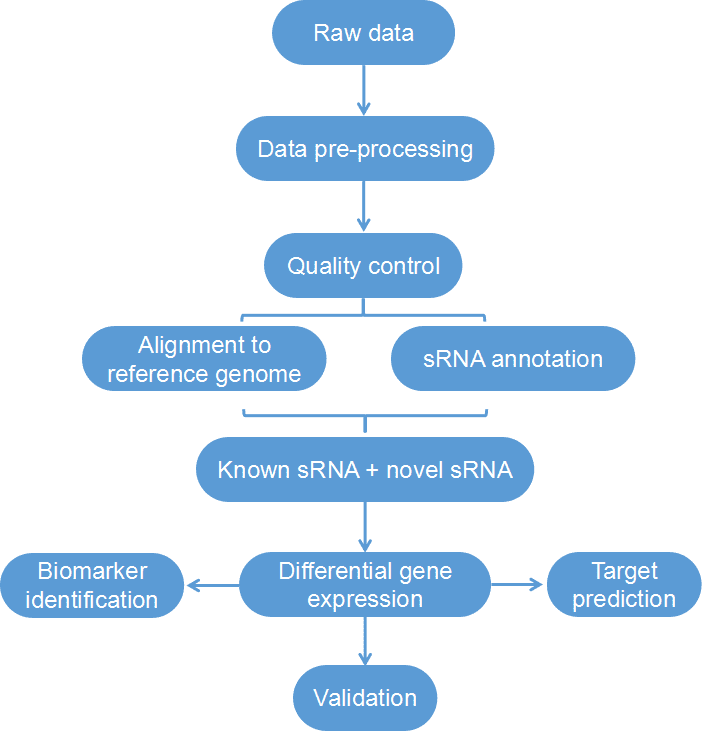
Figure 1. Workflow of bioinformatics analysis of small RNA sequencing.
Table 1. Crucial steps and tools for small RNA sequencing data analysis (Buschmann et al. 2016).
| Step | To consider | Recommended tools or algorithms |
| Data preprocessing | Trimming adapters Removing short reads | Btrim, FASTX-Toolkit |
| Quality control | Library size and read distribution across samples Per base / sequence Phred score Read length distribution Assess degradation Check for over-represented sequences | Btrim, FASTX-Toolkit, FaQCs |
| Read alignment | Reference database or genome Annotation Mismatch rate Handling of multi-reads | Bowtie, BWA, HTSEQ, SAMtools, SOAP2 |
| Normalization | Library sizes and sequencing depth Batch effects Read distribution Replication level Data distribution Replication level | DESeq2, EdgeR, svaseq |
| *DEG analysis | Data distribution Replication level False discovery rate | DESsq2, EdgeR, SAMSeq, voom limma |
| Target prediction | In silico prediction or experimental validation Canonical and non-canonical target regulation | miRanda, miRTarBase, TarBase |
| Biomarker identification | Sensitivity Specificity Classification rate | DESeq2, Simca-Q, Numerous R packages: base, pcaMethods, Mixomics |
* DGE, differential gene expression.
Raw data pre-processing and quality control
To facilitate correct alignments, raw data must be trimmed to accommodate adapter artifacts and sequences with inadequate lengths. Reads less than 16-18 nt representing degraded RNA or adapter dimers need to be removed. Tools such as Btrim, FASTX-Toolkit, FaQCs, and cutadapt are used for this purpose. However, this is not enough for high quality datasets and accurate alignments. There are algorithms such as Quake, ALLPATHSLG, which is dedicated to correcting unreliable base callings by superimposing the most frequent and similar patterns on them. Reads of low quality also need to be removed partially or completely based on their Phred scores. Popular quality trimming algorithms include Cutadapt, Btrim, FASTX Toolkit, FaQCs, and SolexaQA.
After data pre-processing and quality control, the remaining reads should be rid of low quality sequences (quality score < 20) and adapter artifacts, and read lengths should exhibit a distinct peak based on small RNA species of interest (e.g. 21-23 nt for miRNA and 30-32 nt for piRNA).
Small RNA read alignment
Read alignment strategies involve mapping to a reference genome or specific small RNA databases such as mirBase and Rfam. In addition to comparison with specific sequences, homologous datasets from well-studied organisms are also useful due to strong conservation of seed sequences between most small RNA species in different species.
Table 2. The common tools for small RNA sequencing.
| Small RNA read alignment tools | Evaluations or recommendations | |
| algorithm | BLAST aligner, suffix / prefix | Suffix / prefix based on Burrows-Wheeler Transform is fast and efficient in mapping |
| software | Bowtie, BWE, SOAP2 | An evaluation of mapping sensitivity and specificity is strongly recommended. Researches with large datasets or limited time could try BarraCUDA, SOAP3-dp, or MICA. |
Normalization
Systematic variations need to be addressed prior to differential expression analysis. This process is called normalization, which deals with undesired differences between libraries in sequencing depth, GC content, and batch effects. Median normalizing of expression ratios from geometric means has been found to work favorably with diverse kinds of datasets. Zyprich-Walczak et al. (2015) proposed a workflow to determine the most suitable normalization method for a specific dataset.
Differential expression analysis
Differential gene expression (DGE) analysis is vital in small RNA data analysis, which contributes to target prediction and biomarker identification. There are several good tools for this purpose (Table 3), but the optimal tool is highly dependent on the specific dataset.
Table 3. Tools for differential expression analysis of small RNA.
| Tools | Principles | Applicable conditions |
| edgeR | A weighted likelihood approach toward the common dispersion | Appropriate for smaller datasets; Outperforms its competitors for a low number of replicates |
| DESeq | Models the observed mean-variance relationship for all genes via regression | Appropriate for smaller datasets; Outperforms its competitors for experiments with more than 12 replicates |
| SAMSeq | Based on Wilcoxom rank statistics and resampling strategies | Appropriate for datasets with sufficient sample sizes of 10 or more; Low power and specificity for experiments with low sample sizes. |
| NOISeq | Compares the absolute and relative expression differences between and within experimental conditions | Low power and specificity for experiments with low sample sizes. |
| Voom + limma | Voom incorporates the mean-variance trend into a precision weight for each individual normalized observation. Limma includes linear modeling, quantitative weights, and empirical Bayes statistical methods. | Works well with different datasets |
Biomarker identification and target prediction
Biomarker candidates can be identified by differential expression analysis. The tools shown in Table 1 can also be used for biomarker identification. The detected small RNA biomarkers mainly are based on miRNAs. There are several tools and software packages for the in silico functional analysis of miRNA. TargetScan package, TargetFinder and miRanda can be used for in silico target prediction. The predicted targeted genes are further analyzed by Gene Ontology (GO) and KEGG pathway analysis.
Validation
To confirm the small RNA sequencing results, differentially expressed small RNAs need to be examined by qRT-PCR. If it turns out to be consistent with the small RNA sequencing results, the small RNA sequencing data are confidential and reliable. The discovered biomarker signature can, therefore, be assumed after data validation.
Additional Readings:
The challenge and workflow of small RNA sequencing
References:
- Buschmann D., Haberberger A, Kirchner B, et al. Toward reliable biomarker signatures in the age of liquid biopsies-how to standardize the small RNA-Seq workflow[J]. Nucleic acids research, 2016, 44(13): 5995-6018.
- Miao X, Luo Q, Zhao H, et al. Genome-wide analysis of miRNAs in the ovaries of Jining Grey and Laiwu Black goats to explore the regulation of fecundity. Scientific reports, 2016, 6: 37983.
- Zyprych-Walczak J., et al. The impact of normalization methods on RNA-Seq data analysis. Biomed Res. Int., 2015, doi:10.1155/2015/621690.


