
 Sample Submission Guidelines
Sample Submission GuidelinesWhat is Ribo-seq
Ribo-seq, or "Ribosome profiling," refers to the sequencing of actively translating RNA fragments that are associated with ribosomes. This technique provides precise and quantitative information about all translatable molecules in a sample, including mRNAs and other potentially translatable RNA molecules such as lncRNAs and circRNAs.
Ribo-seq is one of the most commonly used methods in translatome sequencing. This technique involves digesting cellular RNA using RNases to obtain ribosome-protected RNA fragments that are actively being translated, often referred to as "ribosome footprints" (RFs). These RFs are typically around 30 nucleotides in length. Subsequently, these ribosome-protected fragments are enriched, deeply sequenced, and analyzed. Ribo-seq provides information at the whole-genome level about protein translation efficiency, enables the discovery of new proteins or short peptides, and allows researchers to calculate the translation efficiency of RNA in conjunction with transcriptome data. By combining this data with proteomics information, the relationship between transient translation and protein accumulation can be analyzed, aiding in the study of protein degradation effects. Translatome sequencing serves as a bridge between RNA and protein, enabling the investigation of various aspects of gene translation, such as levels, regions, and rates. When combined with transcriptome data, small RNA sequencing, proteomics, and other analyses, it facilitates more precise research into post-transcriptional and translational regulation mechanisms.
Our Ribosome-profiling Strategy
CD genomics provides a ribosome-profiling strategy that is based on the deep sequencing of ribosome-protected mRNA fragments and enables genome-wide investigation of translation with sub cordon resolution. Using nuclease digestion, the ribosome position and the translated message can be precisely determined by analyzing the protected ~30-nt area of the mRNA template.
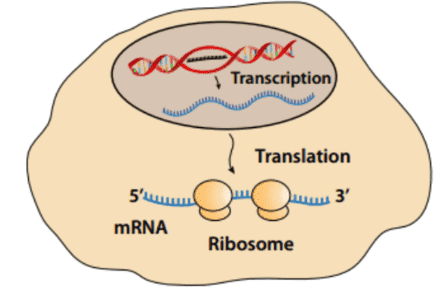
Fig.1 Ribosome-protected mRNA
Application of Ribosome Profiling
- Sequences of ribosome-protected mRNA.
- The study of active mRNA translation with sequencing.
- Prediction of protein abundance.
- Identify translation start sites
- Investigation of transcriptional control and post-transcriptional regulation.
Ribosome Profiling Workflow
Using nuclease digestion to isolate nuclease-resistant ribosome, the translated message can be precisely determined by analyzing the protected 30 nucleotides during translation.

Sample Requirements
- Frozen cell or tissue samples
Sequencing
- Illumina High throughput sequencer
- Flexible service options,single-end or paired-end sequencing, optional reads number according to research goals.
Bioinformatics Analysis
| Basic Analysis | |
| Raw Data Quality Control | |
| Alignment Results Quality Control | |
| Analysis of Read Distribution on the Genome | |
| Gene Coverage Analysis | |
| P-Site Definition Confirmation | |
| ORF Statistics and Corresponding Amino Acid Sequence Analysis | |
| Advanced Analysis | |
| Differential Gene Expression Analysis | |
| gene expression analysis | |
| GO functional analysis | |
| KEGG pathway analysis | |
| Rectome pathway analysis | |
| circRNA translation identification analysis | |
| GO functional analysis of circRNA host genes | |
| KEGG pathway analysis of circRNA host genes | |
| Analysis of circRNA host gene Rectome pathway | |
| Targeted miRNA prediction analysis of circRNA | |
| circRNA and targeted miRNA network regulatory chart | |
| Prediction analysis of circRNA translation potential | |
| Personalized Analysis | |
| Translation Efficiency Analysis of TEs | |
| Analysis of Differential Expression of TE-Related Genes | |
| Clustering Analysis of Differential TE-Related Genes | |
| Enrichment Analysis of Differential TE-Related Genes Using GO and Kyoto Encyclopedia of KEGG Pathways |
Ribo-seq Research Strategy
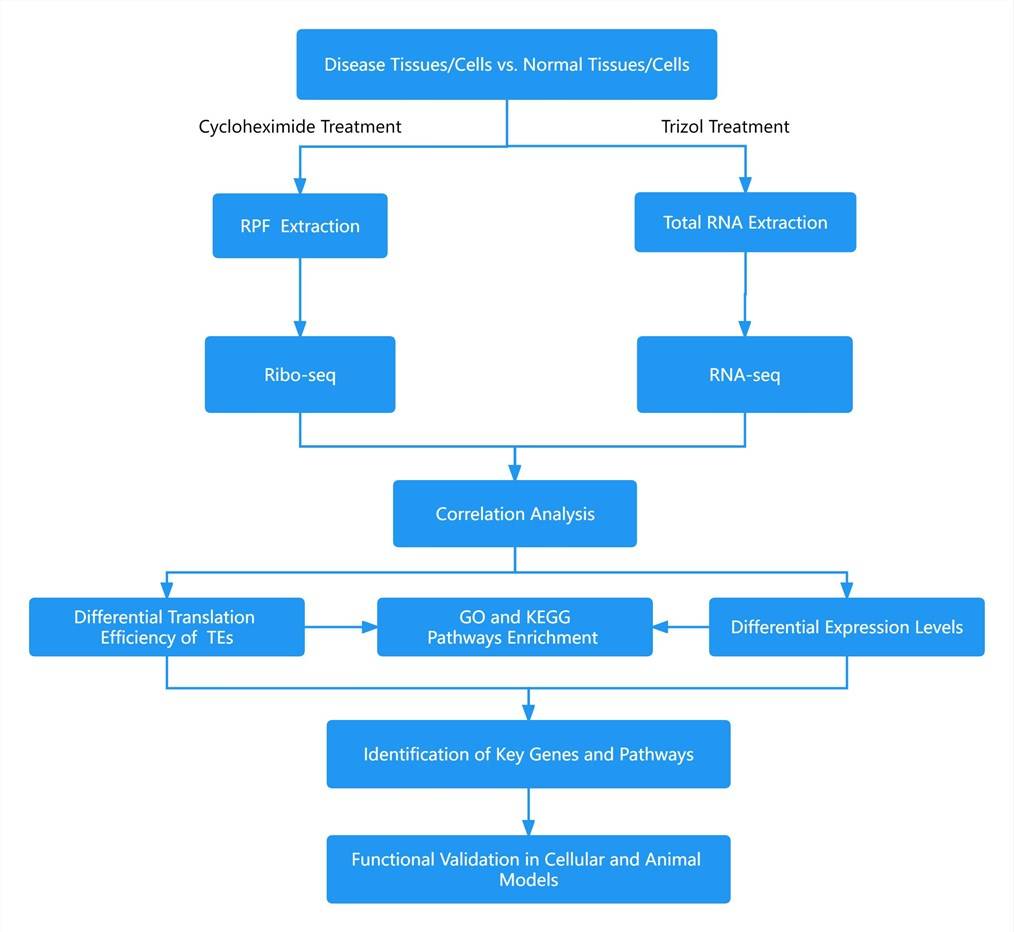
The Translational Landscape of the Human Heart
Impact Factor: 36.216
Sequencing Strategies: Ribo-seq & mRNA-seq
In this study, mRNA sequencing (mRNA-seq) and ribosome profiling (ribo-seq) were conducted on left ventricular myocardial tissues from 65 patients with dilated cardiomyopathy (DCM) and 15 normal controls. The analysis focused on the overall landscape of translated RNA molecules in both DCM and healthy control groups. Combining previous proteomic data on cardiac tissues, this study systematically analyzed translated open reading frames (ORFs) and their corresponding proteins or peptides in left ventricular myocardial tissues. A total of 1,090 upstream ORFs (uORFs), 20,763 conventional ORFs, and 74 downstream ORFs (dORFs) were identified. Additionally, 339 small ORFs (sORFs) from 169 long non-coding RNAs (lncRNAs) were included (see Figure 1). Furthermore, by analyzing ribo-seq data, 40 translatable circular RNA (circRNA) molecules were discovered based on reads crossing the circular RNA junction site, with criteria of a minimum of 9 nt across the junction in at least three samples and a total read count of at least 5 (see Figure 2). These 40 circRNAs originated from 39 genes, including six circRNAs whose translation products were previously hinted at in early proteomic studies. Notably, among these circRNAs, there were well-known molecules such as CDR1as. Importantly, circCFLAR, circSLC8A1, circMYBPC3, and circRYR2 were newly identified circRNA molecules with translational potential in myocardium.
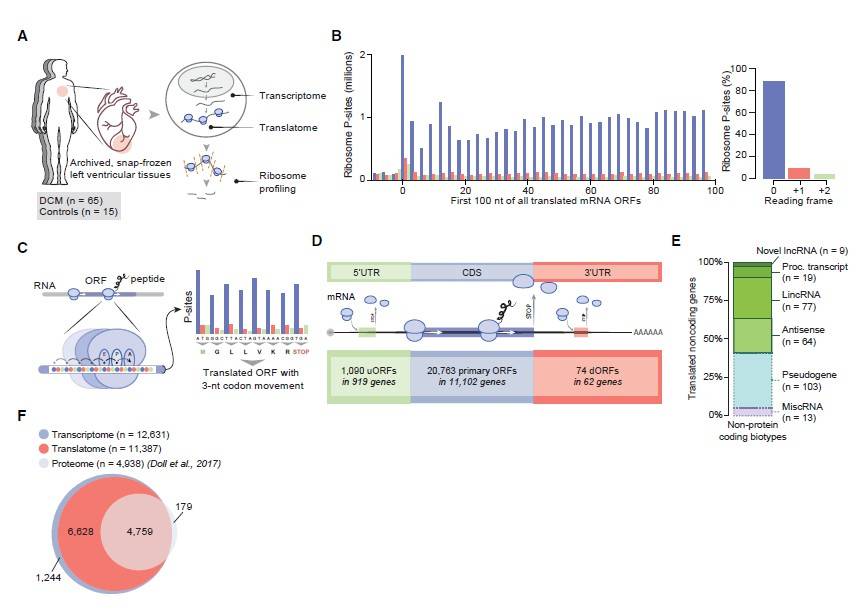 Figure 1. A Snapshot of Active Translation
Figure 1. A Snapshot of Active Translation
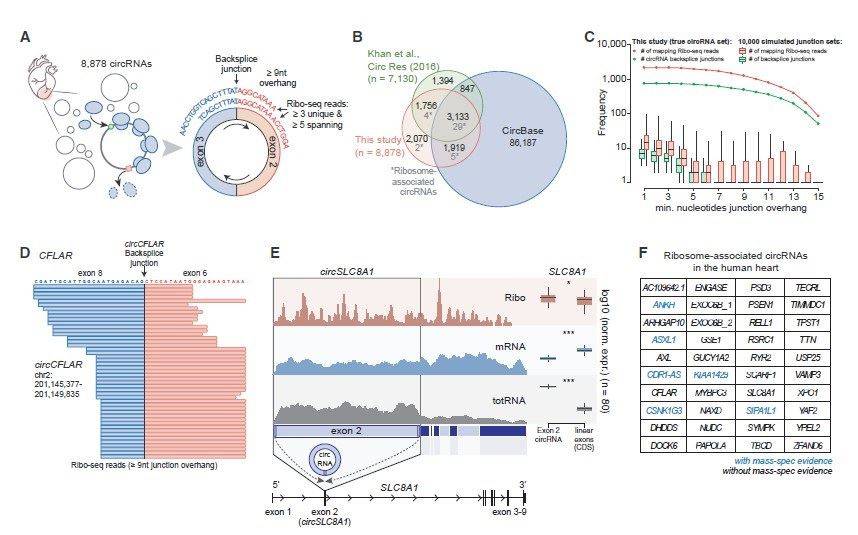 Figure 2. Ribo profiling analysis of translatable circRNA molecules
Figure 2. Ribo profiling analysis of translatable circRNA molecules
Reference:
- van Heesch S, et al,. The Translational Landscape of the Human Heart. Cell. 2019 Jun 27;178(1):242-260.e29. cell.2019


