
 Sample Submission Guidelines
Sample Submission Guidelines
The Application Of Transcriptome In Revealing Disease Mechanism
Transcriptome is a collection of all RNA transcribed by a specific species, tissue or cell under a specific physiological state, including mRNA and non-coding RNA. Transcriptome sequencing can quickly obtain mRNA expression profile; Based on the measured sequence, precise analysis of the sequence and structural information of transcripts such as SNP and variable splicing can be performed simultaneously.
Single cell transcriptome used to reveal tissue specificity
Endothelial cells (EC) exhibit significant functional heterogeneity, depending on the blood vessels and tissues they are located in. Although the transcriptome may bear the imprint of these functional differences, the pathways and networks supporting EC heterogeneity are still not completely clear. Researchers examined transcriptome sequencing data from tissue-specific mouse ECs generated by the Tabula Muris consortium by investigating the transcriptomic basis of EC specificity. Single EC transcriptome data were extracted from 12 organs (adipose tissue, aorta, brain, diaphragm, heart, kidney, liver, lung, breast, pancreas, skeletal muscle and trachea) through annotation results, and there were enough EC for downstream analysis. These cells all have high endothelial gene expression, including Pecam1, Cdh5, Tie1, and Egfl7, with the lowest gene expression in neurons, kidneys, and lung tissues. What is more important, researchers identified novel markers of tissue-specific ECs and signaling pathways that may be involved in maintaining their identity. Sex is an important source of endothelial transcriptome heterogeneity. They found that Lars2 is a gene highly enriched in the endothelial cells of male mice. The results found that markers of heart and lung ECs in mice were conserved in human fetal heart and lung ECs. Additionally, this study identified potential angiocrine interactions between tissue-specific ECs and other cell types by analyzing ligand and receptor expression patterns.
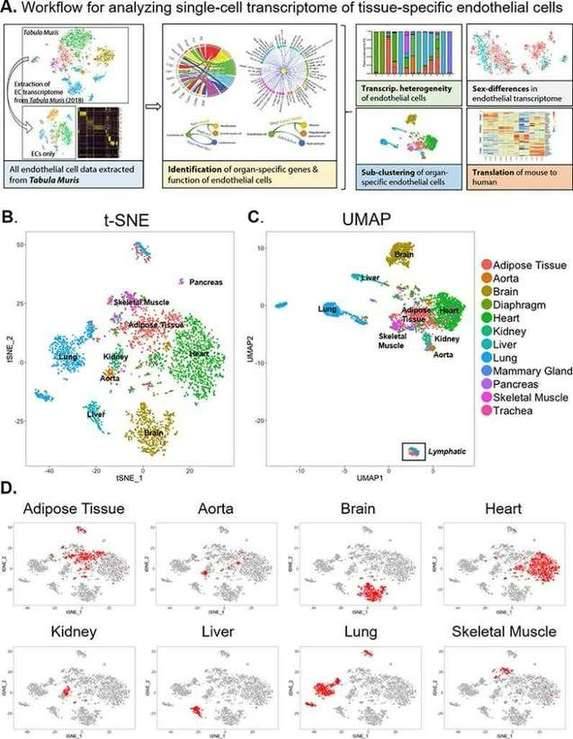 Single-cell transcriptome of endothelial cells in 12 major organs extracted from the Tabula Muris dataset
Single-cell transcriptome of endothelial cells in 12 major organs extracted from the Tabula Muris dataset
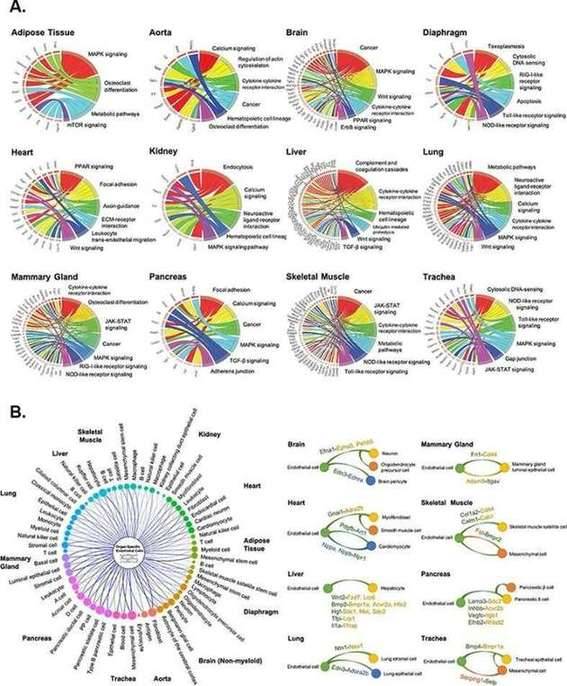 Pathway enrichment and angiocrine relationship prediction analyses
Pathway enrichment and angiocrine relationship prediction analyses
Transcriptome is used to explore the evolutionary mechanism of cancer
Understanding the causes of differences between cancer cells is crucial for understanding tumor evolution, which can provide us with more ideas and treatment strategies. Previous studies have shown that most of these differences originate from the transcriptome. In the mouse model of non-small cell lung cancer (NSCLC), transcriptome plasticity has been proved to be the basis of ITH. Previously, gene expression differences could be obtained using transcriptome of RNA seq data. The transcriptome is implicated as a significant source of phenotypic diversity in analyses of 947 tumor areas, spanning both primary and metastatic illness, together with 96 tumor-adjacent normal tissue samples. During the evolution of tumors, patterns of positive and negative selection are correlated with gene expression levels and ITH. Researchers observe frequent copy number-independent allele-specific expression that is linked to epigenomic dysfunction. Allele-specific expression can also result in genomic-transcriptomic parallel evolution, which converges on cancer gene disruption. The aetiology of single-base RNA alterations is linked to the activity of the RNA-editing enzymes ADAR and APOBEC3A, allowing researchers to identify previously unknown APOBEC activity in malignancies. Researchers combined a number of machine learning techniques to characterize the transcriptome of two primary metastatic tumors. These techniques link the potential for metastatic seeds with the evolutionary context of mutation and proliferation increase in the primary tumor region. These results highlight the interaction between genome and transcriptome in influencing ITH, cancer evolution and metastasis. Overall, this study suggests that these treatments promote tumor evolution and intratumoral heterogeneity. At the same time, it demonstrated the application of detectable circulating tumor DNA in lung cancer detection and identified factors that predict which part of the tumor will lead to recurrence.
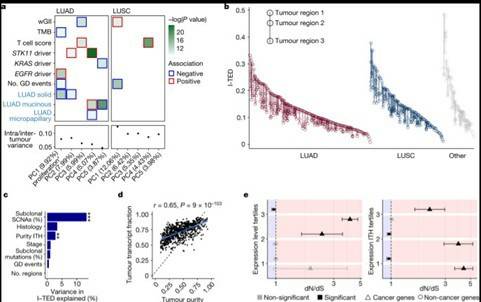 Expression diversity in the TRACERx 421 cohort
Expression diversity in the TRACERx 421 cohort
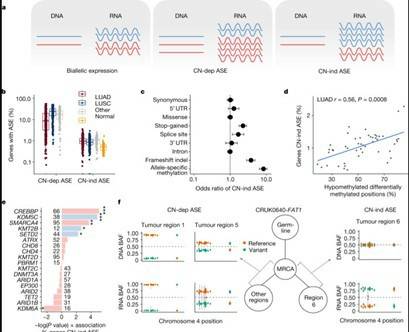 ASE in NSCLC
ASE in NSCLC
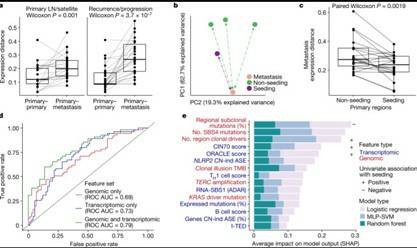 Transcriptional landscape of seeding tumor regions
Transcriptional landscape of seeding tumor regions
So far, transcriptome sequencing and expression profiling is rapidly becoming an irreplaceable method for various studies, including human, animal and plant research, which can more accurately and quickly identify rare and new cells in tissues in an unprecedented way. In addition, with information on gene expression, metabolites, intercellular communication, and spatial landscape at the mRNA and protein levels, it is possible to address the challenges of cell composition and function in health and disease.
References:
- Paik, David T et al. "Single-Cell RNA Sequencing Unveils Unique Transcriptomic Signatures of Organ-Specific Endothelial Cells." Circulation vol. 142,19 (2020): 1848-1862.
- Martínez-Ruiz, Carlos et al. “Genomic-transcriptomic evolution in lung cancer and metastasis." Nature vol. 616,7957 (2023): 543-552.


