
 Sample Submission Guidelines
Sample Submission Guidelines
The Methods of Whole Genome Sequencing
Overview of Whole Genome Sequencing
The genome of each individual organism contains its entire genetic information. Whole genome sequencing technology can comprehensively and accurately analyze entire genomes, thereby breaking the information contained in it and revealing the complexity and diversity of the genome. The emergence of whole genome sequencing technology is a revolutionary advancement in all areas of life sciences. Whole genome sequencing can detect variants, including single-nucleotide variants, insertions/deletions, copy number changes, and large scale structural variants. Whole genome sequencing can be bifurcated into two categories based on the availability of a reference genome: de novo sequencing and resequencing. The presence of a reference genome streamlines and breezes the process of genome assembly.
Differences between WGS and WES
Whole Exome Sequencing (WES) involves utilizing target enrichment techniques to capture and sequence the entire exonic region of the genome. This method can directly detect Single Nucleotide Polymorphisms (SNPs) associated with functional variations in proteins. Within the human genome, despite exons (protein coding regions) comprising only 1% of the genic content, roughly 85% of disease-causing mutations are located in these regions, making WES crucially significant.
Whole Genome Sequencing (WGS), on the other hand, refers to the high-throughput sequencing of the entire genome, analyzing inter-individual variations and annotating SNPs along with genomic structures. Due to the large amounts of comprehensive data that WGS provides, it captures exclusive details that WES or targeted sequencing might overlook. With advances in sequencing technology and substantial reductions in associated costs in recent years, the use of WGS has become increasingly feasible. Furthermore, WGS has the upper hand when it comes to identifying SNPs, insertions, and deletions; hence, it has become an alternative choice for both clinical applications and basic research.
Services you may interested in
Two Classic Approaches for Sequencing Large Genomes
In the early 80s, Sanger successfully completed a whole genome sequencing of the lambda phage by using the shotgun method, and the method was successfully applied to the larger virus DNA, the organelle DNA, and the sequencing of the bacterial genome DNA. Shotgun sequencing is a classic strategy for whole genome sequencing. The shotgun sequencing strategy provides a technical guarantee for large-scale sequencing. The technology first randomly interrupts a complete target sequence into small fragments, sequenced separately, and then splicing them into a consistent sequence by using the overlapping relationships of these small fragments. It mainly includes two methods: one is hierarchical shotgun sequencing (clone-by-clone method) and the other is whole genome shotgun sequencing.
Clone-by-clone sequencing
This method was once adopted by the HGP consortium. This method can generate high density maps, making the genome assembly easier. It generally includes four steps, preparation of BAC clone library, preparation of clone fingerprint, BAC clone sequencing, and sequence assembly. However, this method is time-consuming and costly, so it is seldom used at present.
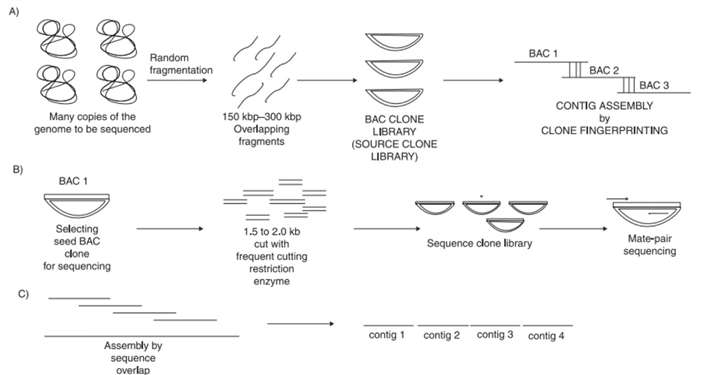 Figure 1. Steps involved in the clone-by-clone sequencing.
Figure 1. Steps involved in the clone-by-clone sequencing.
Whole Genome Shotgun Sequencing (WGS)
WGS generally involves six steps, isolation of genomic DNA, random fragmentation of genomic DNA, size selection using electrophoresis, library construction, paired-end sequencing (PE sequencing), and genome assembly. Two different sizes of DNA fragments including longer insert (2-2.5 kb) and short insert (0.5-1.2 kb) are selected from the agarose gel. While the long inserts are cloned in phage or socmid vectors, the short inserts are cloned in plasmid vectors. The short insert clone library is used for sequencing from both the ends. Since large numbers of clones are sequenced, each of the genomes will be covered more than 10 times. Long insert clones can be used to increase the efficiency of genome assembly.
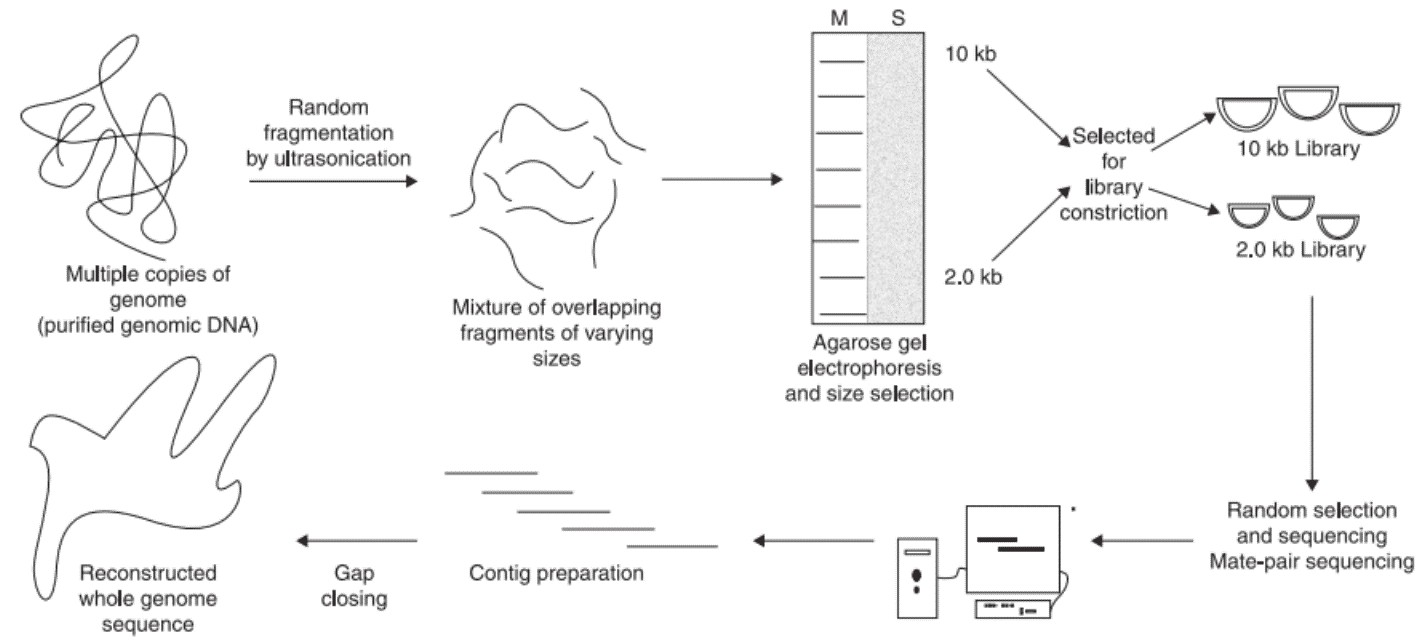 Figure 2. Steps involved in the whole genome shotgun sequencing.
Figure 2. Steps involved in the whole genome shotgun sequencing.
Advantages:
- Does not require genome maps.
- Less time consuming
- Money-saved
Disadvantages:
- Genome assembly for eukaryotic genomes is difficult due to abundant repetitive sequences
- Genome sequencing using this method is not accurate.
NGS Accelerates WGS
Unlike clone-based library approaches, next-generation sequencing platforms utilize a dramatically simplified method of library construction, which has simplified and accelerated the whole genome shotgun sequencing. In generally, genomic DNA is first randomly fragmented using sonication or nebulization, and then are ligated to a platform-specific set of double-stranded adapters to generate a shotgun library. Subsequently, these library fragments can be amplified in situ by hybridization and extension from complementary adapters which are covalently attached to the surface of a glass microfluidic cell or a small bead (depending on the sequencing platform). All NGS instruments utilize a microfluidic device to contain the amplified fragments of the shotgun library, followed by an imaging step that collects data from fragments being actively sequenced.
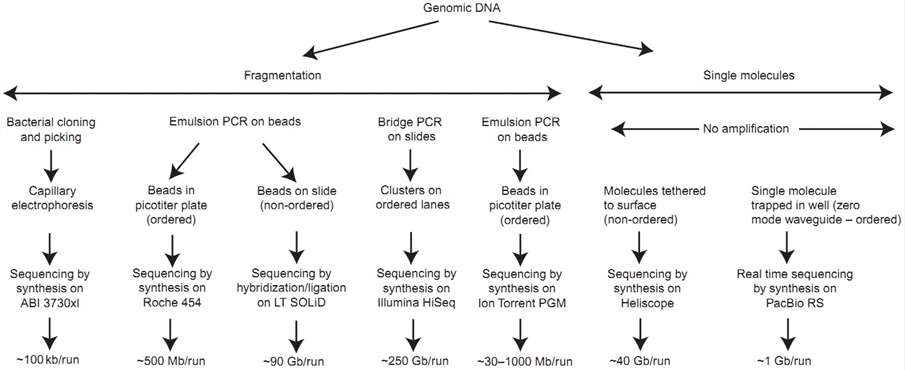 Figure 3. Major steps in employing high-throughput DNA-sequencing methodologies (Ginsburg & Willard 2008).
Figure 3. Major steps in employing high-throughput DNA-sequencing methodologies (Ginsburg & Willard 2008).
WGS Process
We will take the Illumina sequencer as an example to illustrate the workflow of WGS based on high-throughput sequencing.
- Construction of Sequencing Library
The genome is first prepared, and then the DNA is randomly fragmented into hundreds of bases or shorter fragments with specific adapters at both ends. If the transcriptional group is sequenced, the library construction is a bit more troublesome. After the RNA fragmentation, it needs to reverse to cDNA, then add the connector, or reverse the RNA to the cDNA first, then fragment and add the joint. The size of the fragment (insert size) has an impact on the subsequent data analysis and can be selected according to needs. For genome sequencing, several different insert sizes are usually chosen to get more information when assembling.
- Surface Attachment and Bridge Amplification
The reaction of Solexa sequencing is carried out in a glass tube called flow cell, and flow cell is subdivided into 8 Lanes, each of which has a number of fixed single strand joints on the inner surface of each Lane. The DNA fragment of the joint was transformed into a single strand and combined with the primers on the sequencing channel to form a bridge like structure for subsequent preamplification.
- Denaturation and Complete Amplification
The unlabeled dNTP and the common Taq enzyme were added for solid phase bridge PCR amplification, and the single-stranded bridge sample was amplified into a double-stranded bridge fragment. By denaturation, a complementary single strand is released and anchored to the nearby solid surface. By continuously cycling, millions of clusters of double-stranded analytes will be obtained on the solid surface of the Flow cell.
- Single Base Extension and Sequencing
Four fluorescently labeled dNTPs, DNA polymerases, and linker primers were added to the sequenced flow cells for amplification. When each sequencing cluster extends the complementary strand, each fluorescent labeled dNTP is added to release the corresponding fluorescence. The sequencer obtains sequence information of the fragment to be tested by capturing a fluorescent signal and converting the optical signal into a sequencing peak by computer software. The read length is affected by a number of factors that cause signal attenuation, such as incomplete cutting of fluorescent markers. As the length of the reading increases, the error rate will also increase.
- Data Analysis
This step is not strictly a part of the sequencing process, but it only makes sense through the work in front of this step. The raw data obtained by sequencing is a sequence of only a few tens of bases in length, and the contigs that assemble these short sequences through bioinformatics tools are even the framework of the entire genome. Alternatively, these sequences are aligned to an existing genome or a similar species genome sequence, and further analyzed to obtain biologically meaningful results.
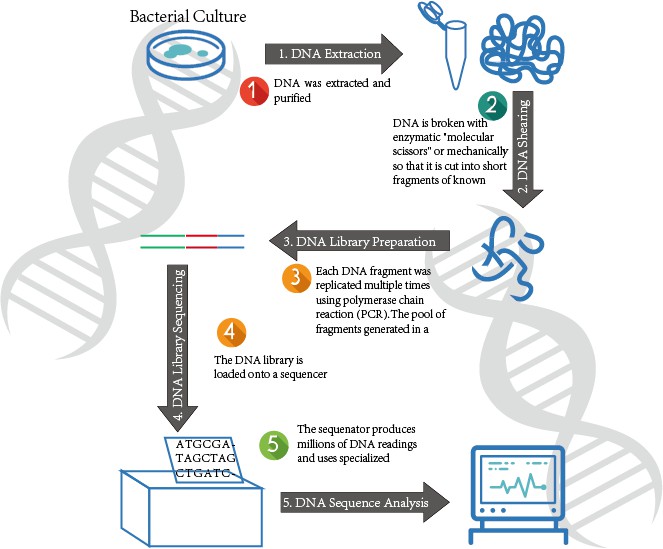 Figure 4. The WGS Process
Figure 4. The WGS Process
Services you may interested in
WGS Sequencing Metrics
- Depth
The sequencing depth, one of the key metrics used to assess volume in genomics, is defined as the ratio of the total recorded base pairs (bp) to the size of the genome. There exists a direct correlation between the sequencing depth and the level of genome coverage, such that an increase in the former contributes to a decrease in false-positive results or sequencing errors. In the context of individual sequencing, efficacious genome coverage and error control can be achieved when employing double-ended or Mate-Pair sequencing strategies, given that the sequencing depth is above the 50X-100X range. This substantial depth consequently facilitates the subsequent assembly of sequences into chromosomes, making the process more efficient and accurate.
- Coverage
Meanwhile, the measure of sequencing coverage pertains to the proportion of the entire genome that is successfully sequenced. This metric is a significant indicator of the randomness involved in sequencing. The relationship between sequencing depth and coverage can be effectively determined through the renowned Lander-Waterman model (1988). According to this model, achieving a sequencing depth of 5X corresponds approximately to the coverage of 99.4% of the entire genome.
Application of WGS
WGS finds its applications across various fields including the determination of mutation rate, genome-wide association studies, medical diagnostics, studies pertinent to rare variations, oncology, epidemiological investigations, and medical genetics, amongst others.
Medical Diagnostic
In the domain of medical diagnostics, in 2009, Illumina, the leading genomics company, introduced its first whole-genome sequencer. This marked a substantial transition as it was approved for clinical use instead of being exclusively utilized for research purposes. In the same year, a team led by Euan Ashley at Stanford University clinically interpreted the complete human genome of bioengineer Stephen Quake, symbolizing the practical establishment of this technology in the field of medical diagnostics.
Medical Genetics
The sphere of medical genetics has also greatly leveraged the cost-effective nature of whole-genome sequencing. WGS is increasingly being employed in deciphering the genetic underpinnings of Mendelian as well as complex diseases, illuminating novel disease biology, and providing substantial assistance in clinical diagnoses and treatment strategies.
Mutation frequencies
WGS facilitates the identification of the mutation rate of the complete human genome. The mutation rate across different human generations (from parents to offspring) stands at approximately 70 new mutations per generation.
Oncology
Within oncology, comprehensive WGS encompasses the reconstruction of subclones based on circulating tumor DNA (ctDNA) in plasma. This paves the way for thorough epigenomic and genomic analyses, revealing the dynamic expression of circulating tumor DNA in every situation.
Epidemiological Investigations
In epidemiological investigations, WGS, having the ultimate discriminative power in differentiating closely-related pathogenic strains, significantly enhances traditional epidemiological investigations of infectious disease outbreaks. By combining WGS with in-depth epidemiological analysis, novel insights have been gained into various aspects. These include the origins and spread dynamics of vast outbreaks caused by Escherichia coli and Vibrio cholerae. Hospital outbreaks induced by Methicillin-resistant Staphylococcus aureus (MRSA), Klebsiella pneumoniae, and Abscessus bacilli have also been investigated. Community-centered outbreak by Mycobacterium tuberculosis and environmental fungal outbreaks associated with natural disasters have received comprehensive analysis due to the integration of WGS.
If you are interested in our genomics services, please feel free to contact our scientists. We are more than happy to be of assistance. In addition to genomics sequencing, we also provide services including transcriptomics, epigenomics, microbial genomics, and PacBio SMRT sequencing.
References:
- Bentley D R. Whole-genome re-sequencing. Current Opinion in Genetics & Development, 2006, 16(6):545-552.
- Fuentespardo A P, Ruzzante D E. Whole-genome sequencing approaches for conservation biology: advantages, limitations, and practical recommendations. Molecular Ecology, 2017, 26(20):5369.
- Batzoglou S, Berger B, Mesirov J, et al. Sequencing a genome by walking with clone-end sequences (abstract): a mathematical analysis// International Conference on Computational Molecular Biology. DBLP, 2000:45.
- Sanger F , Coulson A R, Hong G F, et al. Nucleotide sequence of bacteriophage lambda DNA. Journal of Molecular Biology, 1982, 162(4):729-73.
- Kawarabayasi Y, Sawada M, Horikawa H, et al. Complete sequence and gene organization of the genome of a hyper-thermophilic archaebacterium, Pyrococcus horikoshii OT3. Dna Research, 1998, 5(2):55.
- Kaneko T, Sato S, Kotani H, et al. Sequence analysis of the genome of the unicellular cyanobacterium Synechocystis sp. strain PCC6803. II. Sequence determination of the entire genome and assignment of potential protein-coding regions. Dna Research, 1996, 3(3):185-209.
- Myers E W, Sutton G G, Delcher A L, et al. A Whole-Genome Assembly of. Science, 2014.
- Siegel A F, Engh G V D, Hood L, et al. Modeling the Feasibility of Whole Genome Shotgun Sequencing Using a Pairwise End Strategy. Genomics, 2000, 68(3):237.
- White O, Fraser C M. Genome sequence of the radioresistant bacterium Deinococcus radiodurans R1. Science, 1999, 286(5444):1571-1577.
- May B J, Zhang Q, Li L L, et al. Complete genomic sequence of Pasteurella multocida, Pm70. Proceedings of the National Academy of Sciences of the United States of America, 2001, 98(6):3460-3465.
- Ginsburg G S, Willard H F. Genomic and personalized medicine. Academic Press, 2008.
- Ormond K E, Wheeler M T, Hudgins L, et al. Challenges in the clinical application of whole-genome sequencing. The Lancet, 2010, 375(9727): 1749-1751.
- Le V T M, Diep B A. Selected insights from application of whole-genome sequencing for outbreak investigations. Current opinion in critical care, 2013, 19(5): 432-439.
- Wu J, Wu M, Chen T, et al. Whole genome sequencing and its applications in medical genetics. Quantitative Biology, 2016, 4(2): 115-128.
- Ashley E A, Butte A J, Wheeler M T, et al. Clinical assessment incorporating a personal genome. The Lancet, 2010, 375(9725): 1525-1535.
- Roach JC, Glusman G, Smit AF, et al. Analysis of genetic inheritance in a family quartet by whole-genome sequencing. Science, 2010, 328 (5978): 636–9.
- Campbell CD, Chong JX, Malig M; et al. Estimating the human mutation rate using autozygosity in a founder population. Nat. Genet, 2012, 44 (11): 1277–81.
- Herberts Cameron, Annala Matti, Sipola Joonatan, et al. Deep whole-genome ctDNA chronology of treatment-resistant prostate cancer. Nature, 2022, 608 (7921): 199–208.