
 Sample Submission Guidelines
Sample Submission Guidelines
Principles and Workflow of 16S/18S/ITS Amplicon Sequencing
16S/18S/ITS amplification sequencing uses the next/third generation sequencing platform and performs high throughput sequencing of PCR products from specific regions such as 16S rDNA/18S rDNA/ITS/ functional genes. It overcomes the disadvantage of some microorganisms that is difficult or impossible to culture, and obtains the information of microbial community structure, evolutionary relationships and microbial correlation with environment in environmental samples.
What Is 16S rDNA /18S rDNA/ITS?
16s rDNA: 16S rDNA is a DNA sequence encoding small subunit rRNA of prokaryotes with a length of about 1542bp. With a moderate molecular size and low mutation rate, 16S rDNA is the most commonly used marker in the study of bacterial systematics. The 16S rDNA sequence consists of 9 variable regions and 10 conservative regions, the conserved region sequences reflect the genetic relationships between species, while the variable region sequences reflect the difference between species. 16S rDNA sequencing is mainly used to analyze the diversity of bacteria or archaea.
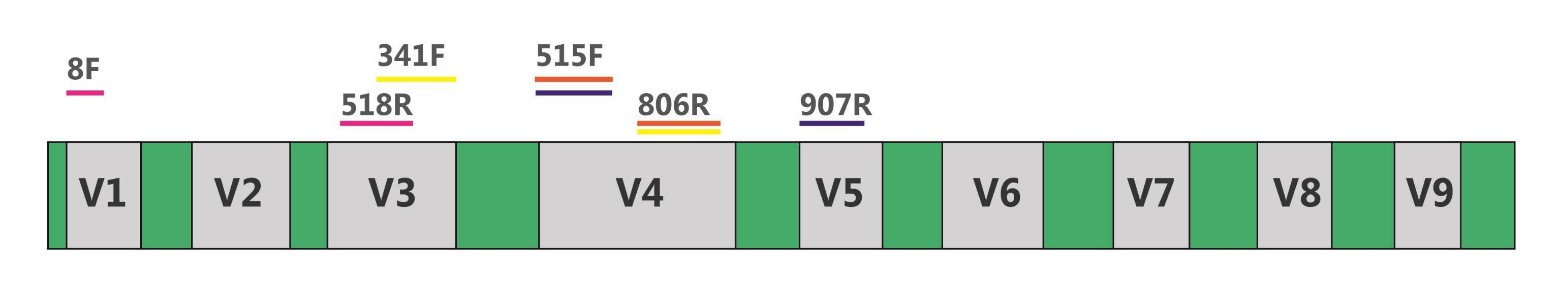 Fig.1 16S rDNA and amplification primers
Fig.1 16S rDNA and amplification primers
18S rDNA: 18S rDNA is a DNA sequence encoding small subunit rRNA of eukaryotic ribosomes. Like 16S rDNA, 18S rDNA sequence also consists of conservative regions and variable regions (V1-V9, absence of V6). Among variable regions, V4 has the most complete database information and the best classification effect, it is the mostly used and the best choice for 18S rRNA gene analysis notes. 18S rDNA sequencing reflects the species differences among eukaryotic organisms in given samples.
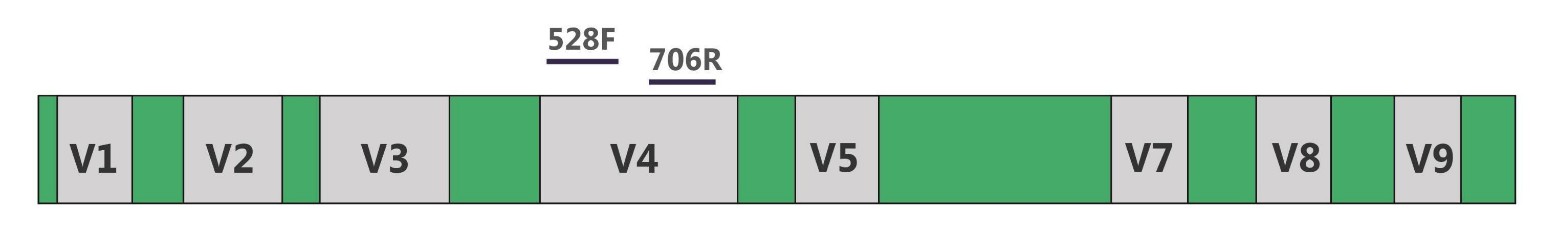 Fig.2 18S rDNA and amplification primers
Fig.2 18S rDNA and amplification primers
ITS: ITS (Internal Transcribed Spacer) is part of the non-transcriptional region of the fungal rRNA gene. The ITS sequences used for fungal identification usually include ITS1 and ITS2. Because in fungi, 5.8S, 18S, and 28S rRNA genes are highly conserved, whereas ITS can tolerate more mutations in the evolutionary process due to less natural selection pressure, and exhibits extremely wide sequence polymorphism in most eukaryotes. At the same time, the conservative type of ITS is relatively consistent within species, and the differences between species (or ever stains) are obvious. ITS sequence fragments are small (350 bp and 400 bp in length, respectively) and easy to analyze. They have been widely used in phylogenetic analysis of different fungi.
 Fig.3 ITS and amplification primers
Fig.3 ITS and amplification primers
Services you may interested in
What Is 16S/18S/ITS amplicon sequencing?
The methodological principle of amplified fragment sequencing employs Polymerase Chain Reaction (PCR) technology to selectively amplify particular target DNA fragments. Generally, this pertains to the 16S rRNA gene regions of bacteria and archaea or the 18S rRNA/ITS regions for eukaryotes. These fragments embody highly-conserved sequence regions whilst concurrently encompassing sufficient variation zones, rendering them competent instruments for the discernment and differentiation of various microbes.
16S/18S/ITS amplicon sequencing uses Illumina or PacBio sequencing to read the PCR products which are amplified with suitable universal primers of one or several regions of 16S/18S/ITS. By detecting the sequence variation and abundance of the target area, the information of species classification and abundance, population structure, phylogenetic evolution and community comparison of environmental samples could be obtained. Investigating microbial diversity bears significant theoretical and practical implications for understanding relationships between microbes and the environment, environmental management, as well as harnessing microbial resources.
| Sequencing Methods | Research Subjects |
| 16S rDNA/rRNA sequencing | Bacteria or Archaea |
| 18S rDNA/rRNA sequencing | Eukaryotes |
| ITS sequencing | Fungi |
Advantages of 16S/18S/ITS Amplicon Sequencing
Efficiency in Identification: Compared to traditional methods of identification such as cloning or culturing, sequencing microbial communities of 16s/18s/ITS offers a faster and more accurate approach.
Speed: Contrasting traditional methods of microbial classification and identification, amplicon sequencing generates considerable data in a relatively short timeframe, thereby accelerating the processing and analysis of microbial samples.
High Sensitivity: Amplicon sequencing can detect low abundance species within microbial communities, even those comprising a minimal proportion within the community can be effectively identified.
Diversity: Amplicon sequencing facilitates simultaneous analysis of multiple microbial communities, encompassing a wide range of microbes such as bacteria, archaea, and fungi, thereby promoting its widespread applicability.
Double Region Detection: This approach offers flexibility in targeting one or more variable regions, allowing for longer sequence reads and more precise analysis of colonies.
Lower Costs: Amplicon sequencing requires less depth when compared to metagenomic sequencing, hence offering better cost-effectiveness.
Quantifiable: Amplicon sequencing allows for quantitative analysis of sequencing data, determining the relative or absolute abundance of microbes, thereby enabling a quantitative assessment of microbial community composition and changes.
Workflow of 16S/18S/ITS Amplicon Sequencing
The principal steps of 16S/18S/ITS amplicon sequencing include extraction of total DNA from samples, PCR amplification of target areas, library construction, sequencing, and bioinformatics analysis.
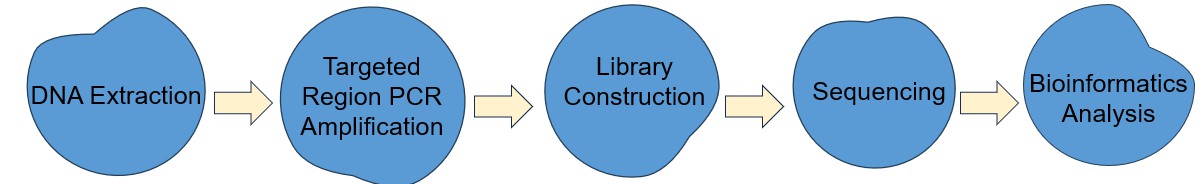 Fig.4 The workflow of 16S/18S/ITS amplicon sequencing
Fig.4 The workflow of 16S/18S/ITS amplicon sequencing
DNA Extraction: This marks the process of obtaining total DNA from a given sample. The extracted DNA embraces segments hailing from various microorganisms, encompassing areas of the targeted gene.
Targeted Region PCR Amplification: The primary elements consist of primer design, PCR amplification, purification of its products, and the final appraisal of the purity of these products. The design of the primer should tightly and specifically integrate with the sequence located within the region of interest, often devised in a conservative area, to assure the specificity of amplification. The PCR amplification involves the preparation of the corresponding mixture, the optimization of PCR reaction conditions, and then subjecting the mixture for the PCR amplification reaction.
The quintessential PCR protocol encompasses a series of cycles, including denaturation, annealing, and elongation stages, enabling the primer to bind with the DNA template and generate new DNA strands. The byproducts of the PCR reaction necessitate purification via a PCR purification kit, removing unreacted primers, dNTPs, and other impurities. Eventually, gel electrophoresis is employed to inspect the size and purity of the amplified products, thereby confirming the presence of only the amplification products of the target region.
Library Construction: We recommend the fusion primer library construction method, that is, the primers fused with the target sequence primers and the adapter, index and other sequences are synthesized in advance, then the genomic DNA targets are directly amplified by PCR. Amplicon libraries are purified and an equimolar pool of the amplicon libraries is prepared. The dilution required for template preparation is determined and followed by sequencing.
Sequencing: The current sequencing platforms mainly include Illumina Miseq/HiSeq and third-generation sequencing platform.
- Illumina NGS (MiSeq/HiSeq2500/HiSeq4000): Due to the limitation of reading length, the NGS platform can only select a single variable region, double variable regions or triple variable regions as the target regions for the sequencing. When sequencing, only the completely sequenced Reads (Tags) can be used for further analysis, so different amplification regions should strictly follow the corresponding sequencing strategy. For example, if you chose V4 for analysis, the PE250 sequencing is needed, but for V1-V3 regions, the sequencing strategy should be PE300. Only in this way can the completeness of sequences be ensured. The original data is filtered out to remove low-quality reads and leave high-quality clean data for later analysis.
- PacBio SMRT Sequencing: Unlike NGS, the third generation sequencing platform can carry out full-length sequencing for 16S/18S/ITS, and it's sequence alignment rate and identification accuracy rate are higher than that of the NGS.
Bioinformatics Analysis: Reads are spliced into Tags according to the Overlap relationship between reads, and tags are aggregated into OTUs with a given similarity, and then OTUs are annotated by comparing OTUs with databases.
Operational taxonomic units (OTUs) are often used to classify groups of closely related individuals. In general, if the similarity of different 16S rDNA/18S rDNA/ITS sequences is higher than 97%, those sequences can be defined as an OTU. Each OTU corresponds to a different 16S rDNA/18S rDNA/ITS sequence, that is, each OTU corresponds to one species. By OTU analysis, the microbial diversity and the abundance of different microorganisms in the sample can be known.
Then based on OTU and species annotation results, sample species complexity analysis and species difference analysis are conducted. Species based analysis, LDA effect size analysis and more analysis are provided too.
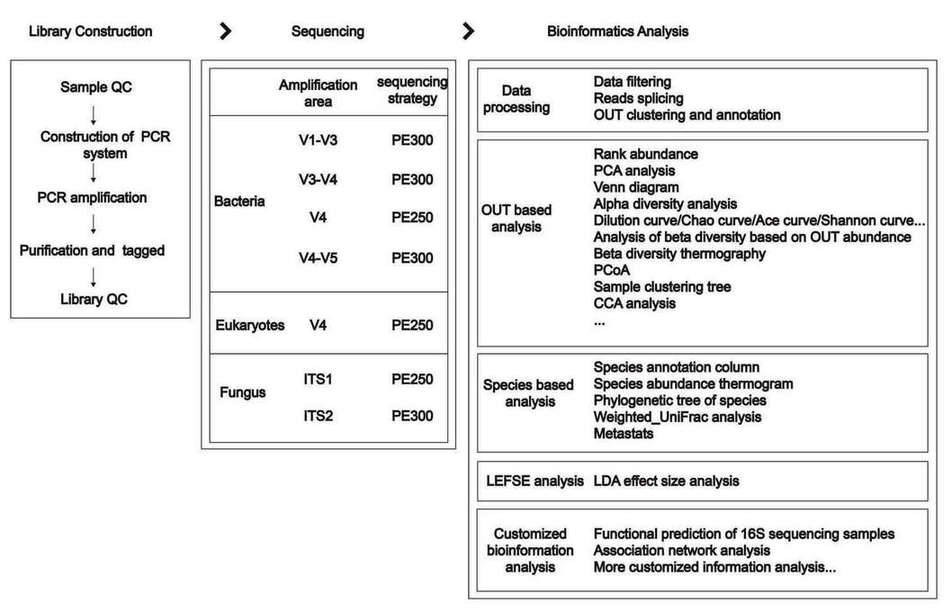 Fig.5 The key workflow of 16S/18S/ITS amplicon sequencing
Fig.5 The key workflow of 16S/18S/ITS amplicon sequencing
Application of 16S/18S/ITS Amplicon Sequencing
The utilization of 16S/18S/ITS amplicon sequencing spans multiple sectors including biology, medicine, and more. These sequences, including 16S rRNA, 18S rRNA, and ITS, operate as molecular fingerprints of organisms such as bacteria, archaea, and fungi, thus reflecting the composition and structure of microbial communities. The process of amplicon sequencing on these sequences allows one to understand the diversity, abundance, and distribution characteristics of microbiota within varying environmental samples, such as soil, aquatic habitats, gut flora, etc. This consequently reveals the structure and evolutionary tendencies of microbial communities. Furthermore, 16S/18S/ITS amplicon sequencing provides a vital tool for the classification and identification of microbes. By comparing amplicon sequences with known sequences in databases, unknown microbes can be categorized and their taxonomic status and phylogenetic relationships can be determined. This bears significant relevance for new species identification, species typing of environmental microbes, and detection of pathogenic microbes.
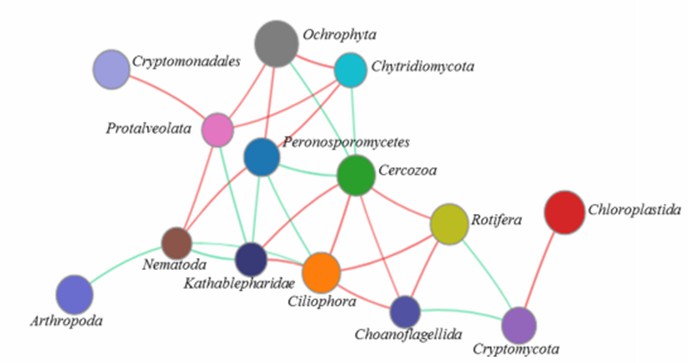 Figure 6. Network analysis of the microeukaryotic community at the phylum level. (Xu et al., 2020)
Figure 6. Network analysis of the microeukaryotic community at the phylum level. (Xu et al., 2020)
Microorganisms play pivotal roles in the biosphere, participating in the cycling of matter and the flow of energy, enhancing soil fertility, promoting plant health, and functioning within animal intestines. By employing 16S/18S/ITS amplicon sequencing, we can explore the functional traits of microbial communities, such as those involved in nitrogen and carbon cycles, and bioluminescence synthesis, thereby deepening our understanding of the impact and roles of microbiomes within ecosystems.
In disease diagnostics and treatment, the microbial community is intricately intertwined with host health. 16S/18S/ITS amplicon sequencing can therefore serve as a tool for disease diagnosis and therapy. For instance, an imbalance in gut microbiota is associated with conditions such as inflammatory bowel disease and autoimmune diseases. Amplicon sequencing of gut microbiota can facilitate an understanding of these microbial shifts, providing critical reference points for disease diagnosis and treatment strategies. In biotechnology and bioengineering, microorganisms are extensively utilized in processes such as fermentative production, environmental remediation, and biodegradation. Through amplicon sequencing of microbiomes, we can isolate strains with particular functions, thereby paving the way for the development of efficient biological technologies and engineering applications.
Differences Between 16S Sequencing and Metagenomic Sequencing:
Sequencing Principles:
- 16S Sequencing involves amplifying specific variable regions (e.g., V3-V4 or V4) of microbial genomic DNA through PCR after extraction, followed by library construction and sequencing.
- Metagenomic Sequencing randomly fragments microbial genomic DNA into small segments of 300-500bp, adds sequencing adapters to both ends of the fragments, and then proceeds with sequencing.
Varied Taxonomic Identification Depth:
- Sequences obtained from 16S sequencing often cannot be annotated to the species level, whereas metagenomic sequencing can identify microorganisms down to the species or even strain level.
Research Objectives:
- 16S Sequencing primarily investigates species composition, evolutionary relationships among species, and the diversity of microbial communities.
- Metagenomic Sequencing, building upon 16S sequencing analysis, allows for in-depth exploration of genetic and functional aspects (e.g., Gene Ontology, Pathway analysis).
At CD Genomics, our expert team with extensive experience can help you fully understand microbial communities and take advantage of them. In addition to 16S/18S/ITS Amplicon Sequencing, we also provide other microbial genomics services, including:
Metagenomic Shotgun Sequencing
Viral Metagenomic Sequencing
Metatranscriptomic Sequencing
Microbial Whole Genome Sequencing
Viral Genome Sequencing
References:
- Michelsen, C. F., Pedas, P., Glaring, M. A., et al. (2014) 'Bacterial diversity in greenlandic soils as affected by potato cropping and inorganic versus organic fertilization', Polar Biology, 37(1), 61-71.
- Edwards, J., Johnson, C., Santosmedellí, C., Lurie, E., Podishetty, N. K., & Bhatnagar, S., et al. (2015) 'Structure, variation, and assembly of the root-associated microbiomes of rice', Proceedings of the National Academy of Sciences of the United States of America, 112(8), E911.
- Evans, C. C., Lepard, K. J., Kwak, J. W., Stancukas, M. C., Laskowski, S., & Dougherty, J., et al. (2014) 'Exercise prevents weight gain and alters the gut microbiota in a mouse model of high fat diet-induced obesity', Plos One, 9(3), e92193.
- Shehab, N., Li, D., Amy, G. L., Logan, B. E., & Saikaly, P. E. (2013) 'Characterization of bacterial and archaeal communities in air-cathode microbial fuel cells, open circuit and sealed-off reactors', Appl Microbiol Biotechnol, 97(22), 9885-9895.
- Man, K. C., Au, C. H., Chu, K. H., Kwan, H. S., & Chong, K. W. (2010) 'Composition and genetic diversity of picoeukaryotes in subtropical coastal waters as revealed by 454 pyrosequencing', Isme Journal, 4(8), 1053.
- Lie, A. A. Y., Liu, Z., Hu, S. K., Jones, A. C., Kim, D. Y., & Countway, P. D., et al. (2014) 'Investigating microbial eukaryotic diversity from a global census: insights from a comparison of pyrotag and full-length sequences of 18s rrna genes', Appl Environ Microbiol, 80(14), 4363-4373.
- Lu, L., Yin, S., Liu, X., Zhang, W., Gu, T., & Shen, Q., et al. (2013) 'Fungal networks in yield-invigorating and -debilitating soils induced by prolonged potato monoculture', Soil Biology & Biochemistry, 65, 186-194.
- Orgiazzi, A., Lumini, E., Nilsson, R. H., Girlanda, M., Vizzini, A., & Bonfante, P., et al. (2012) 'Unravelling soil fungal communities from different mediterranean land-use backgrounds', Plos One, 7(4), e34847.
- Lakshmanan, V., Ray, P., & Craven, K. D. (2017) 'Rhizosphere sampling protocols for microbiome (16s/18s/its rrna) library preparation and enrichment for the isolation of drought tolerance-promoting microbes', Methods Mol Biol, 1631, 349-362.
- Xu H, Zhang S, Ma G, et al. 18S rRNA gene sequencing reveals significant influence of anthropogenic effects on microeukaryote diversity and composition along a river-to-estuary gradient ecosystem. Science of the total environment, 2020, 705: 135910.


