
 Sample Submission Guidelines
Sample Submission Guidelines
What is Telomere-To-Telomere Sequencing?
What is Telomere?
Telomeres are crucial structural elements located at the terminal ends of linear chromosomes in eukaryotic organisms. These specialized regions play a vital role in maintaining the integrity and stability of chromosomes. Comprising simple yet highly repetitive DNA sequences, telomeric DNA poses challenges for assembly due to its intricate nature.
Numerous research findings underscore the significance of telomeres in cellular dynamics. With each cell division in new cells, the telomere at the chromosomal end undergoes shortening. Once this shortening reaches a critical point, the cell loses its ability to divide further. Consequently, scientists refer to telomeres as the "clock of life," recognizing their pivotal role in determining the lifespan of cells.
CD Genomics undertakes further optimization by filling in assembled gaps, identifying telomeres and filaments, ultimately culminating in the generation of a Haplotype-Resolved T2T genome.
What is the Telomere-to-Telomere (T2T) Genome?
The advent of long-read sequencing technology, particularly the powerful combination of high-precision PacBio HiFi sequencing and the extended continuity offered by ONT ultra-long sequencing, has successfully addressed the challenging assembly issues associated with mitotic or highly repetitive genomic regions. This breakthrough has significantly enhanced the continuity and integrity of chromosomes, laying the groundwork for the creation of Telomere-to-Telomere (T2T) genomes.
Essentially, a T2T genome aims to achieve a high-quality genomic sequence that spans from one telomere to the other, characterized by exceptional accuracy, continuity, and integrity.
A notable milestone in this endeavor is the release of the human T2T-CHM13 de novo genome, which effectively fills in critical gaps in the genomic landscape.
Please read our article The Complete T2T Genome by Sequencing Opens The Door to Post-genomic Era for more information.
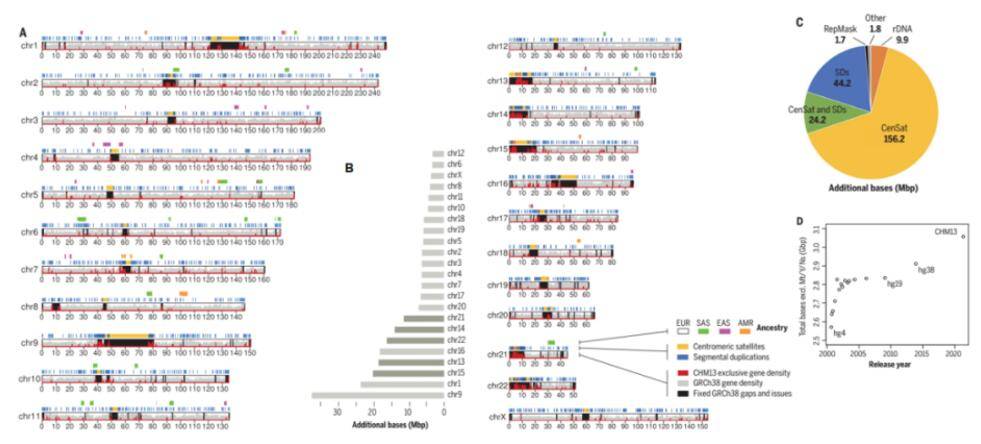 The complete T2T-CHM13 human genome assembly (Nurk S et al., 2022)
The complete T2T-CHM13 human genome assembly (Nurk S et al., 2022)
This includes the comprehensive representation of mitotic satellite arrays, the proximal repeat region, and the short arms of the five telocentric chromosomes. Unlocking these intricate genomic regions is instrumental for conducting mutational and functional studies. The completed map of the human genome resulting from this effort incorporates the addition or correction of 238 Mb of sequence, with 182 Mb being entirely new, and the annotation of 2,226 new genes. Consequently, this refinement has led to the elimination of tens of thousands of false-positive variants in each sample, notably reducing false positives by over 90% in 269 medically relevant gene tests.
Detailed analysis of mitogen-associated sequences has unveiled a compelling correlation between the mitogen's location and the evolution of layered repeat amplification in its surrounding DNA. Furthermore, comparisons of X chromosome mitoplasts from different individuals reveal a notable degree of structural, epigenetic, and sequence variation within these complex and rapidly evolving genomic regions.
We are also closely following the advancements in T2T genome research for other species. To learn more, please read our articles.
- The Complete Rice Nipponbare Reference Genome
- Fungal Genome De Novo Sequencing Promotes The Discovery And Research of Plant Pathogenic Fungi
- Human Pangenome Reference Consortium Releases Data from 30 Genomes
Why is T2T Genome Assembly So Challenging?
The intricacies of T2T genome assembly stem from the intricate nature of genomic structures, prompting a reliance on three generations of sequencing technology—specifically, PacBio HiFi, ONT ultra-long, and Hi-C sequencing technologies. The integration of Hi-C technology is crucial for acquiring the relative position information of genes on the chromosome, facilitating the completion of genome-level assembly at the chromosomal level. In navigating complex regions, manual adjustments with extensive assembly experience become imperative, ultimately yielding a high-quality T2T reference genome sequence.
Despite these advancements, significant challenges persist, particularly in reading through lengthy repetitive and mitotic regions in certain species. In the case of the new human genome results, the strategy involves avoiding the sequencing of two distinct X chromosomes in normal human cells. Instead, the complexity associated with assembling two haplotypes of a diploid genome is sidestepped by employing a haploid cell line derived from a human gravid, featuring two identical X chromosomes.
Please refer to our article Successful Decoding of the Y Chromosome: A Milestone in Human Genome Unraveling for more information.
The direct mapping of highly duplicated chromosome regions in diploid normal humans necessitates further investigation and more comprehensive assemblies. This is especially true for other species less studied than the human genome, where assembling mitotic grains and addressing assembly gaps induced by highly repetitive regions become even more formidable challenges. Consequently, obtaining a complete and high-quality T2T genome for a species remains a formidable task.
Haplotype-Resolved T2T Genome Assembly
Current genome assembly methods commonly overlook distinctions between homologous chromosomes, leading to the creation of chimeric sequences amalgamated from both parental chromosomes.
For specific diploid species, such as hybrids or those characterized by high genome heterozygosity, and polyploid species, employing haplotype genome assembly becomes essential. This approach allows for the extraction of genetic information from both parental sources, enabling the study of the asymmetric evolution of subgenomes and the exploration of expression differences among haplotype alleles. This nuanced understanding sets the stage for more robust investigations into resequencing, trait localization, evolution, gene function, gene editing, and other subsequent research endeavors.
CD Genomics T2T Haplotype Genome Sequencing Service
Leveraging PacBio HiFi sequencing data, CD Genomics employs Hi-C and Nanopore ultra-long sequencing to facilitate haplotype typing. Employing HIC and kmer clustering, we meticulously exclude chimeric assemblies, ensuring the accurate disassembly of the species' haplotype genome. CD Genomics undertakes further optimization by filling in assembled gaps, identifying telomeres and filaments, ultimately culminating in the generation of a Haplotype-Resolved T2T genome.
To gauge the precision of genome assembly, a comprehensive assessment of assembly quality, specifically haplotype splitting accuracy, is imperative upon completion. This includes subphaser validation, evaluation of haplotype genome typing accuracy (switch error), BUSCO integrity assessment, and covariance analysis among haplotypes. This process employs a flexible and adaptable assembly method complemented by a meticulous assessment protocol for haplotype genomes.
Additionally, a rigorous assembly quality assessment is conducted to evaluate the accuracy of gap filling post-genome typing. This involves scrutinizing assembly continuity (number of gaps per chromosome), consistency (comparison rate of NGS and long-read sequencing data), completeness (BUSCO assessment), accuracy (QV values of the entire genome and each chromosome), telomere and filament sequence identification, and covariance comparison analysis with historically assembled versions of the same species. This comprehensive evaluation ensures the reliability and precision of our gap-filling processes in the context of haplotype genomes.
Application of T2T Haplotype Genome Sequencing Services
Discover the versatility of our T2T Haplotype Genome Sequencing Services in diverse scenarios, including subgenome comparisons, studies on specific insertions/deletions, gene count variations, differential methylation levels, variations in Gene networks, and the origin and evolution of polyploid species.
- Unlocking the Potential for Downstream Molecular Breeding Guidance
Our T2T Haplotype Genome Sequencing Services empower precise genomic editing for complex species, providing invaluable insights for downstream molecular breeding initiatives.
- Delving into Hybrid Vigor Theories
Explore and understand the theories behind hybrid vigor, unraveling the genetic intricacies of mixed-species advantages through our comprehensive haplotype genome sequencing.
- Investigate Allele-Specific Expression Patterns (ASE)
Our services facilitate in-depth studies of allele-specific expression patterns, shedding light on the unique regulatory mechanisms governing gene expression.
- Comparative Selection Pressure Analysis (Ka/Ks)
Evaluate selection pressures by comparing Ka/Ks ratios, gaining critical insights into the evolutionary forces shaping genetic diversity within complex species.
- Subgenome Comparisons
Uncover the nuances of subgenome variations through meticulous comparisons, providing a comprehensive understanding of genomic structures.
- Exploring Specific Insertions/Deletions and Other Mutations
Our services offer a detailed examination of specific insertions, deletions, and other mutations, unraveling the genetic variations that contribute to species diversity.
- Comparative Gene Count Analysis
Analyze differences in gene counts among diverse species, gaining insights into the genetic factors that contribute to species-specific traits and characteristics.
- Differential Methylation Level Comparisons
Explore variations in DNA methylation levels, elucidating epigenetic differences that play a pivotal role in gene regulation and expression.
- Gene Network Regulation Differences
Understand differences in gene network regulation through comparative analysis, providing valuable information on the complex interplay of genes within a species.
- Polyploid Origin and Evolution
Our services delve into the origin and evolutionary history of polyploid species, unraveling the intricate genetic events that have shaped their existence.
- Ancestral Source and Evolutionary Trajectory
Trace the ancestral sources and evolutionary trajectories of complex species, gaining insights into their evolutionary history and genetic heritage.
Reference:
- Nurk S, Koren S, Rhie A, et al. The complete sequence of a human genome. Science, 2022, 376(6588): 44-53.