
 Sample Submission Guidelines
Sample Submission Guidelines
Overview of cDNA Synthesis
What is cDNA synthesis
cDNA synthesis, achieved through the process of reverse transcription, involves transcribing RNA into DNA, thereby serving as a vital amendment and supplementation to the central dogma of molecular biology. Within experimental contexts, two distinct modes of expression typically emerge: reverse transcription and retrotranscription. The former entails the deliberate extraction of RNA for the purpose of synthesizing DNA, while the latter denotes the autonomous process by which RNA viruses transcribe RNA into DNA. Notably, the former occurs ex vivo, whereas the latter transpires in vivo.
The synthesis of cDNA encompasses both first-strand and second-strand synthesis. Generally, methods for synthesizing the first strand of cDNA involve utilizing total RNA or purified mRNA as templates, catalyzing synthesis with RNA-dependent DNA polymerase (reverse transcriptase), often employing Oligo (dT) primers or random primers. Subsequently, second-strand synthesis may be performed to yield complete double-stranded cDNA.
Detailed elucidation of the methods, influencing factors, experimental procedures, precautions, and applications of cDNA synthesis within a scholarly article assumes paramount importance. Such elucidation aids readers in comprehensively understanding the principles and methodologies underlying this technique, as well as its practical utility across various domains of research.
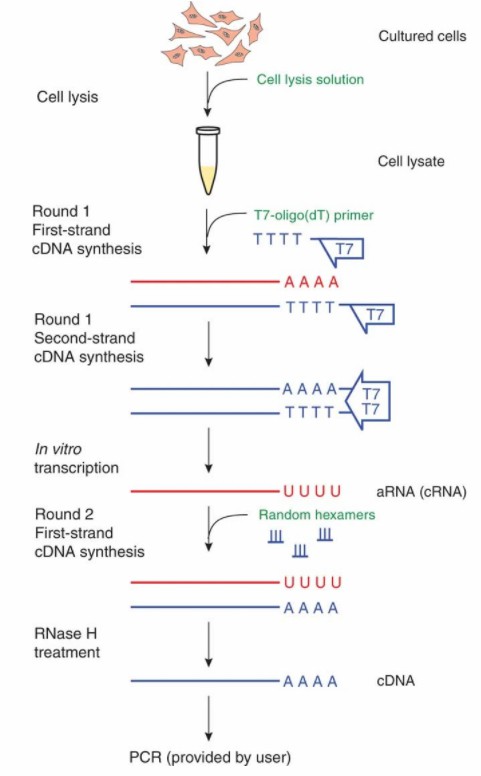 An overview of the procedure for the cDNA Synthesis from Cell Lysates. (Judith E Meis et al,. Nature Methods 2009)
An overview of the procedure for the cDNA Synthesis from Cell Lysates. (Judith E Meis et al,. Nature Methods 2009)
Methods of cDNA Synthesis
| Method | Principle | Characteristics |
| Self-priming Method | In this approach, the synthesized single-stranded cDNA forms a short hairpin structure at the 3' end, providing a ready-made primer for the synthesis of the second strand. Subsequent to denaturation of the DNA-RNA hybrid strand generated in the first-strand synthesis reaction, the second strand of cDNA is synthesized using either Escherichia coli DNA polymerase I Klenow fragment or reverse transcriptase. Finally, digestion of the loop with the single-strand-specific S1 nuclease facilitates further cloning. | - Difficult to control - Predisposed to sequence loss or rearrangement |
| Template-switching Method | This method relies on the presence of dNTPs to induce RNAse H cleavage and gapping on the mRNA strand of the hybrid chain, generating a series of RNA primers for second-strand synthesis. These RNA primers serve as primers for the synthesis of the second strand under the action of Escherichia coli DNA polymerase I. | - Highly effective - Direct utilization of first-strand reaction products without purification - Eliminates the need for S1 nuclease digestion to cleave single-stranded hairpin loops. |
Factors Influencing cDNA Synthesis
The process of reverse transcription, although seemingly straightforward, is subject to various influencing factors, including template quality, primer design, enzyme activity, and reaction conditions.
Template
RNA Secondary Structure (High GC Content, Complex Folding)
RNA molecules fold into intricate secondary structures due to their sequence composition, presenting various motifs such as helices, loops, bulges, internal loops, and multi-branch loops. The stability of these structures varies depending on the base pairing; for instance, GC base pairs are more stable due to the presence of three hydrogen bonds, while AU base pairs involve two hydrogen bonds and GU pairs are the least stable with only one hydrogen bond. Consequently, RNA templates with higher GC content tend to exhibit more complex secondary structures, potentially impeding primer extension during reverse transcription and affecting the length of synthesized cDNA. To mitigate this, denaturation of secondary structures can be achieved by incubating the template at 65°C for 5 minutes followed by rapid cooling on ice. Additionally, conducting the reaction at elevated temperatures (e.g., 55°C) can reduce the formation of RNA secondary structures.
Purity and Concentration Determination
The purity of RNA significantly influences reverse transcription, as contaminants introduced during RNA purification, such as salts, metal ions, ethanol, and phenol, commonly act as inhibitors of reverse transcriptase. Nanodrop spectrophotometry is typically employed to assess RNA purity, with optimal values falling within the range of 1.8 < OD260/OD280 < 2.0 (values below 1.8 indicate protein or phenol contamination, which can be addressed by phenol extraction; values above 2.0 may suggest residual guanidine thiocyanate). Additionally, the OD260/OD230 ratio should exceed 2 (values below 2 indicate the presence of guanidine thiocyanate, β-mercaptoethanol, or ethanol residues, necessitating repeated ethanol precipitation and washing).
Integrity
The integrity of RNA significantly impacts the length and quality of synthesized cDNA. Special precautions, such as wearing gloves and masks, and using RNA-free consumables throughout RNA extraction, handling, and storage processes are crucial to preserving RNA integrity. RNA integrity directly affects the length of synthesized cDNA, thereby influencing downstream cloning experiments and the accuracy of quantitative gene expression assays.
Removal of Genomic DNA (gDNA)
The presence of gDNA in total RNA can lead to false-positive results in downstream qPCR experiments, as gDNA can serve as a template for PCR amplification. Thus, prior to reverse transcription, it is imperative to remove gDNA from the RNA template to ensure that only cDNA is synthesized. DNase I treatment is commonly employed for gDNA removal; however, it is essential to inactivate DNase I before reverse transcription to prevent its degradation activity on cDNA. While heat inactivation of DNase I can degrade RNA, and addition of EDTA as an inactivator introduces new RNAse inhibitors, careful optimization is required to maintain RNA integrity while ensuring complete gDNA removal.
Primer Selection in cDNA Synthesis
Oligo(dT) Primers
Oligo(dT) primers consist of approximately 12-18 repeated oligothymidine nucleotides, specifically designed to anneal to the poly(A) tail of eukaryotic mRNA. Consequently, they are unsuitable for RNA lacking a poly(A) tail structure, such as prokaryotic RNA or microRNA, and are not recommended for use with degraded RNA samples, such as those from formalin-fixed paraffin-embedded (FFPE) tissues.
Eukaryotic mRNA typically constitutes only 1%-5% of total RNA. Therefore, Oligo(dT) primers are preferentially chosen for constructing cDNA libraries from eukaryotic sources or for cloning full-length cDNA and performing 3' RACE experiments. However, for complex RNA templates with extensive secondary structures, primer extension may prematurely terminate upon encountering these structures, potentially resulting in loss of information from the mRNA 5' end when using Oligo(dT) primers.
Random N6-N9 Primers
Random primers consist of short oligonucleotides composed of random nucleotide sequences, typically comprising 6 nucleotides, known as random hexamers. These primers lack specificity and can anneal to various RNA species, including rRNA, tRNA, non-coding RNA, small RNA, degraded RNA, and RNAs with complex secondary structures. While random primers can enhance cDNA yield and concentration, they may lead to shorter cDNA synthesis products.
Gene-Specific Primers
Gene-specific primers (GSPs) are designed to specifically complement the target mRNA sequence, resulting in highly targeted cDNA synthesis, particularly suitable for subsequent PCR amplification of known target sequences. These primers are commonly employed in one-step RT-PCR reactions. In cases where GSPs fail to efficiently synthesize the first-strand cDNA, Oligo(dT) or random primers may serve as alternatives.
Enzymes in cDNA Synthesis
Different reverse transcriptases exhibit varying functional activities, demonstrating diverse efficiencies in cDNA synthesis, including length, yield, and adaptability to complex templates.
RNA-dependent DNA Polymerase Activity: Catalyzes the synthesis of DNA from dNTPs using RNA as a template.
RNaseH Activity: Cleaves RNA within RNA-DNA hybrid molecules during the reaction. Reducing this activity prevents degradation of the RNA template before synthesizing longer cDNA molecules.
DNA-dependent DNA Polymerase Activity: Uses the first synthesized cDNA single strand as a template to generate the second cDNA strand.
Thermal Stability: This property is crucial for cDNA synthesis. Elevating the temperature during reverse transcription helps denature high-GC or structurally complex RNA templates, facilitating the synthesis of full-length cDNA.
Fidelity: Refers to the accuracy of sequence replication during RNA reverse transcription into cDNA. Since reverse transcriptases lack 3'-5' exonuclease activity and error-correction mechanisms, the error rate in synthesized cDNA is relatively high. Generally, except in cases where high sequencing accuracy is required, errors introduced during reverse transcription are negligible, as they are not amplified in subsequent steps.
Inhibitor Resistance: Robust reverse transcriptases are essential for successful cDNA synthesis when templates have low purity or contain inhibitors.
Reaction Process
The optimal temperature for reverse transcription typically stands at 42°C. However, in cases involving templates with high GC content or complexity, elevating the reverse transcription temperature to 50°C might prove beneficial. The resulting products of reverse transcription can be promptly utilized in qPCR reactions or stored short-term at -20°C. For prolonged preservation, it is recommended to aliquot and store them at -80°C to avoid detrimental freeze-thaw cycles.
In essence, achieving an efficient reverse transcription reaction hinges upon several factors: the quality of RNA templates, the suitability of reverse transcriptase enzymes, the selection of appropriate primers, and the establishment of optimal reaction conditions. It is imperative to meticulously consider all these influencing factors throughout the experimental process to synthesize high-fidelity cDNA suitable for subsequent applications.
cDNA Synthesis Principle
cDNA synthesis entails the conversion of mRNA into cDNA through a series of orchestrated biochemical steps:
mRNA Extraction: Total RNA, harboring mRNA molecules, is meticulously extracted from cellular sources.
Reverse Transcription Reaction: The extracted mRNA serves as a template and undergoes reaction with reverse transcriptase enzyme (typically reverse transcriptase reverse transcriptase), catalyzing the formation of single-stranded cDNA.
Reverse Transcriptase Synthesis of cDNA: Leveraging the mRNA template's poly(A) tail region with poly(T) primers, in conjunction with the reverse transcriptase's reverse transcription activity, single-stranded cDNA is meticulously synthesized.
DNA Synthesis: DNA polymerase (such as DNA polymerase I) is harnessed for the synthesis of the complementary strand, leading to the formation of double-stranded cDNA. This process necessitates DNA polymerase I buffer, three dNTPs, and oligonucleotide primers.
Enzymatic Cleavage: The resultant double-stranded cDNA is subjected to precise enzymatic cleavage using specific enzymes to excise RNA templates or other non-cDNA sequences.
Ligation: Adapter molecules are strategically ligated to the termini of the cDNA, facilitating subsequent amplification and cloning endeavors.
In essence, cDNA synthesis entails the reverse transcription of mRNA into single-stranded cDNA, followed by the DNA polymerase-mediated synthesis of complementary strands, enzymatic cleavage, and ligase-mediated concatenation, culminating in the production of cDNA.
cDNA Synthesis Protocol
cDNA First-Strand Synthesis:
1. In a sterile, RNA-free, 0.2 mL microcentrifuge tube, combine the following reaction mixture:
Template RNA: Total RNA 1-5 μg or poly(A+) mRNA 0.1-0.5 μg
Primers: Oligo(dT)18 (0.5 μg/μL) 1 μL, or Random Hexamer Primers (0.2 μg/μL) 1 μL, or Sequence-specific Primers 20 pmol.
RNase-free ddH2O: Bring to a final volume of 17 μL.
2. Gently mix and centrifuge for 3-5 s.
3. Heat the reaction mixture in a 70°C water bath for 5 min, followed by incubation on ice for 30 s, then centrifuge for 3-5 s.
4. Keep the tube on ice and add the following components:
cDNA First-Strand Buffer (5×): 4 μL
RNase Inhibitor (20 U/μL): 1 μL
dNTP Mix (10 mmol/L): 2 μL
5. Gently mix and centrifuge for 3-5 s.
6. Incubate the reaction mixture at 37°C for 5 min, then add 1 μL of M-MLV Reverse Transcriptase (20 U/μL) to achieve a final volume of 25 μL. Transfer 5 μL to another tube and add 2-5 μCi [α-32P] dCTP (>400 Ci/mmol) for radioactivity measurement of the first-strand isotope incorporation.
7. Incubate the reaction mixture at 37°C for 60 min (if using Random Hexamer Primers, pre-incubate at 25°C for 10 min, then continue at 37°C for 60 min).
8. Terminate the reaction by heating at 70°C for 10 min, then cool on ice for subsequent cDNA second-strand synthesis or store frozen.
9. Stop the reaction in the tubes designated for incorporation measurement by adding 95 μL of 50 mmol/L EDTA, bringing the total volume to 100 μL. Take 90 μL for gel electrophoresis analysis (preceded by phenol extraction) and reserve 10 μL for isotope incorporation radioactivity measurement.
cDNA Second-Strand Synthesis:
1. Second-strand synthesis can be directly performed in the reaction mixture from the first-strand synthesis. Take 20 μL of the first-strand reaction mixture and sequentially add:
- 10× Second-Strand Buffer: 10 μL
- DNA Polymerase I: 23 U
- RNase H: 0.8 U
- RNase-free ddH2O to a final volume of 100 μL.
2. Gently mix. If isotopic labeling for radioactivity measurement of the second strand is desired, transfer 10 μL to another tube and add 2-5 μCi [α-32P] dCTP.
3. Incubate at 14°C for 2 hours (extend to 3-4 hours for synthesis of cDNA longer than 3 kb).
4. For incorporation measurement, add 90 μL of 50 mmol/L EDTA to the designated tube, reserve 10 μL for isotopic labeling radioactivity measurement, and use the remaining for gel electrophoresis analysis.
5. Heat the reaction mixture from the cDNA second-strand synthesis tube at 70°C for 10 minutes, then cool on ice after low-speed centrifugation.
6. Add 2 U of T4 DNA Polymerase and incubate at 37°C for 10 minutes.
7. Terminate the reaction by adding 10 μL of 200 mmol/L EDTA.
8. Perform phenol/chloroform extraction on the cDNA reaction mixture, followed by centrifugation at 12,000 rpm for 5 minutes.
9. Transfer the aqueous phase to another tube, add 1/10 volume of 3 mol/L NaAc (pH 5.2), mix, then add 2.5 times the volume of cold ethanol (-20°C) and incubate at -20°C for 1 hour.
10. Centrifuge at 4°C, 12,000 rpm for 15 minutes, carefully discard the supernatant, and wash the pellet with 70% ethanol.
11. Centrifuge at 4°C, 12,000 rpm for 5 minutes, discard the supernatant, and air-dry at room temperature.
12. Dissolve the pellet in 10-20 μL of TE buffer.
cDNA Synthesis Product Quantification:
1. Take 3 μL of each reaction mixture containing isotopes and spot them on glass fiber filter paper. Allow them to air dry at room temperature; these samples represent total radioactive activity.
2. Similarly, take 3 μL of each reaction mixture and mix it with 100 μL of salmon sperm DNA (1 mg/mL). After thorough mixing, add 0.5 mL of 5% TCA and vortex. Incubate the mixture on ice for 5-30 minutes.
3. Filter the reaction mixture through glass fiber filter paper, wash with cold 5% TCA three times (each time with 5 mL TCA), followed by a rinse with 5 mL anhydrous ethanol. These samples represent spiked radioactive activity.
4. Measure the intensities of both total radioactive activity and spiked radioactive activity using a Geiger counter or liquid scintillation counter.
5. First-strand cDNA synthesis product quantification:
- First-strand spike-in rate (%) = [spiked radioactive activity (cpm) / total radioactive activity (cpm)] × 100%
- Spiked dNTPs (nmol) = 2 nmol dNTP/μL × reaction volume (μL) × (first-strand spike-in rate)
- Synthesized cDNA quantity (ng) = spiked dNTP (nmol) × 330 ng/nmol
- mRNA to cDNA conversion rate = [synthesized cDNA quantity (ng) / template RNA quantity (ng)] × 100%
For instance, if the total radioactive activity intensity is 254,000 cpm, the spiked radioactive activity intensity is 3,040 cpm, and 1 μg of RNA template is used with a reaction volume of 25 μL, then:
- Spike-in rate = (3,040/254,000) × 100% = 1.2%
- Spiked dNTP quantity = 2 nmol dNTP/μL × 25 × 1.2% = 0.6 nmol
- Synthesized cDNA quantity = 0.6 nmol dNTP × 330 ng/nmol = 198 ng
- mRNA to cDNA conversion rate = (198 ng / 1000 ng) × 100% = 19.8%
Considering that 20% (5 μL/25 μL) of 1000 ng RNA is used for spiking, and the reaction volume comprises 80% of the total volume, the actual first-strand cDNA synthesis quantity is 0.8 × 198 ng = 158 ng.
6. Second-strand cDNA synthesis product quantification:
- Apart from removing first-strand spiked dNTPs, the method is similar to first-strand yield calculation.
Second-strand spike-in rate = [spiked radioactive activity (cpm) / total radioactive activity (cpm)] × 100% Spiked dNTP quantity (nmol) = [(0.4 nmol/μL × reaction volume (μL)) - first-strand spike-in nmol] × second-strand spike-in rate Second-strand cDNA synthesis quantity (ng) = nmol dNTP × 330 ng/nmol Double-strand cDNA conversion rate = [second-strand cDNA synthesis quantity (ng) / first-strand cDNA synthesis quantity (ng)] × 100%
For example, if the second-strand spiked radioactive activity intensity is 2,780 cpm, and the total radioactive activity intensity is 235,000 cpm:
- Second-strand spike-in rate = (2,780/235,000) × 100% = 1.18%
- Spiked dNTP quantity = [(0.4 nmol/μL × 100 μL) - 0.48 nmol] × 1.18% = 0.47 nmol
- Synthesized second-strand cDNA quantity (ng) = 0.47 nmol × 330 ng/nmol = 155 ng
- Double-strand cDNA conversion rate = (155 ng / 158 ng) × 100% = 98%
Generally, a first-strand conversion rate of 12%-50% and a double-strand conversion rate of 50%-200% are considered optimal.
cDNA Synthesis Product Electrophoresis Analysis
Isotopically labeled cDNA products can be analyzed using alkaline agarose gel electrophoresis. Both the synthesized products and DNA molecular weight standards should be labeled with 32P. The DNA 5'-end labeling kit can be used for labeling.
1. Prepare a 1.4% alkaline agarose gel in alkaline gel electrophoresis buffer and equilibrate it in alkaline electrophoresis buffer for at least 30 minutes.
2.Adjust the volume of the sample solution with TE buffer to match the volume of the first and second-strand yield determination solutions. Add an equal volume of 2x loading buffer (2x loading buffer should be stored at -20°C or added before each use).
3.Load the samples onto the gel, ensuring that the dye front is approximately 1/3 of the gel length from the front edge. Apply an electric field with a voltage of 5 V/cm.
4.Stop the electrophoresis and immerse the gel in 7% TCA at room temperature for 30 minutes (or until the dye changes from blue to yellow). Remove the gel and place it on filter paper, then air-dry for several hours.
5.Wrap the dried gel in plastic wrap and expose it to X-ray film at room temperature (or at -70°C if using an intensifying screen).
Considerations for cDNA Synthesis
Selection of appropriate reverse transcriptase: Different reverse transcriptases possess distinct characteristics and suitability criteria. For instance, for library construction and cloning of unknown sequences, a reverse transcriptase with terminal transferase activity and template switching capability, such as MMLV RTase, may be required. Conversely, for shorter transcripts like those used in qPCR cDNA preparation, AMV RTase might be more suitable.
Elimination of genomic DNA contamination: Prior to reverse transcription, treat RNA samples with DNaseI to remove any contamination from genomic DNA. Ensure complete inactivation of DNaseI to prevent degradation of cDNA in subsequent steps.
Choice of suitable primers: Select primers based on the experimental objectives. For example, oligo-dT primers are typically used for eukaryotic mRNA, while random primers should be used for prokaryotic RNA or RNA lacking polyadenylation.
Ensure quality of RNA template: High-quality RNA template is crucial for synthesizing full-length cDNA. Perform gel electrophoresis to assess RNA integrity and ensure purity, avoiding repeated freeze-thaw cycles.
Control of reaction conditions: Appropriate reverse transcription temperature and time are vital for enhancing the efficiency and quality of cDNA synthesis. For RNA templates with complex secondary structures or high GC content, it may be necessary to increase the reverse transcription temperature or use specialized reverse transcriptases.
Storage of cDNA: Reverse transcription products can be stored short-term at -20°C, but long-term storage is recommended by aliquoting and storing at -80°C.
Additionally, for specific experimental needs such as GeneRacer assays, new techniques involving dephosphorylation, decapping, RNA adaptor ligation followed by reverse transcription may be necessary, requiring mRNA with cap and Poly A structures. During cDNA synthesis, attention should also be paid to avoiding RNA template degradation and erroneous introduction of cDNA, while enhancing cDNA fidelity and resistance to inhibition.
cDNA Synthesis Applications
The process of cDNA synthesis has a range of applications in lab workflows, such as sequencing, qRT-PCR, gene expression studies, cDNA library preparation, gene cloning, and gene discovery studies.
cDNA Synthesis for RNA Sequencing
cDNA synthesis, a critical component of genetic expression analysis and transcriptomics research, plays a fundamental role in converting RNA molecules into cDNA. Following the outlined procedures and application recommendations allows researchers to obtain high-quality cDNA suitable for downstream applications such as RNA sequencing.
At CD Genomics, we offer high-quality RNA sequencing services to assist researchers in obtaining accurate and reliable transcriptome data. Our RNA sequencing services cover various aspects, including whole transcriptome sequencing, targeted sequencing, single-cell sequencing, among others, providing researchers with comprehensive solutions for transcriptomic studies.
You may interested in
Learn More
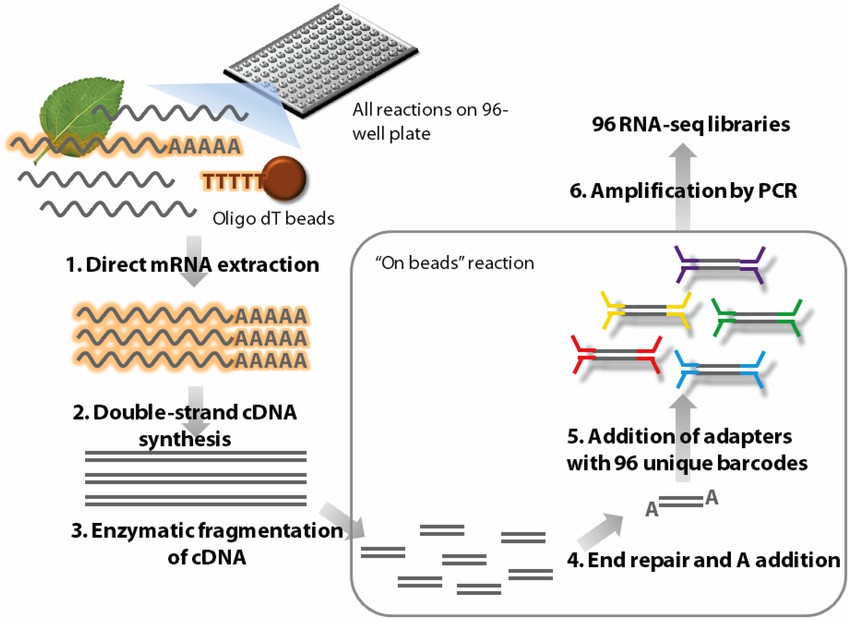 Outline of the high-throughput RNA-seq (HTR) library preparation. (Ravi Kumar et al,. Frontiers in Plant Science 2012)
Outline of the high-throughput RNA-seq (HTR) library preparation. (Ravi Kumar et al,. Frontiers in Plant Science 2012)
Gene Discovery and Expression Profiling
cDNA synthesis is a foundational process in gene discovery endeavors, facilitating the exploration of the complex realm of gene expression. Through the synthesis of cDNA from mRNA templates, researchers delve into the varied patterns of gene expression across different physiological contexts. This methodology not only assists in uncovering new genes but also elucidates their regulatory mechanisms and functional significance within biological systems.
Utilization of cDNA Microarray Technique
A notable application of cDNA synthesis pertains to the field of cDNA microarray technology. This advanced method enables extensive gene expression profiling, enabling the simultaneous examination of thousands of genes. Through the hybridization of cDNA probes originating from diverse experimental conditions onto microarray platforms, researchers can attain comprehensive understandings of the molecular signatures linked to various diseases and biological phenomena.
Gene Cloning and Recombinant Protein Production
The synthesis of cDNA assumes a paramount significance extending far beyond the mere discovery of genes. In the realm of gene cloning initiatives, cDNA synthesis establishes itself as a steadfast and dependably effective methodology for generating recombinant proteins. Through the incorporation of cDNA fragments into sophisticated expression vector systems, researchers are equipped with the capabilities to induce the expression of proteins of particular interest within a heterologous host environment. These host systems could be as varied as standard bacterial strains or yeast cultures. Such a maneuver facilitates the cultivation of substantial quantities of immaculately pure proteins. Notably, the magnitude of this process has direct implications in conducting intricate biochemical and functional examinations as well as the development of therapeutic modalities.
Leveraging cDNA Libraries for Functional Genomics
cDNA libraries, composed of cloned cDNA fragments representing the expressed genes within a cell or tissue, serve as invaluable resources for functional genomics investigations. Through the screening of cDNA libraries, researchers can pinpoint genes linked to specific cellular processes or phenotypes of interest. Moreover, cDNA libraries facilitate the functional elucidation of genes via gain- or loss-of-function methodologies, offering insights into their functions in both physiological and pathological contexts.
Molecular Epidemiology and Pathogen Characterization
In infectious disease research, cDNA synthesis occupies a pivotal position in molecular epidemiology investigations and pathogen characterization. Through the synthesis of cDNA from viral RNA genomes, researchers can trace the dissemination of viral pathogens and unravel their evolutionary dynamics. Furthermore, cDNA synthesis expedites the characterization of viral genomes, thereby facilitating the creation of diagnostic assays and targeted therapeutic interventions.
Advancements in Rapid cDNA Synthesis for Virus Genomics
Recent advancements in cDNA synthesis techniques have brought about a paradigm shift in virus genomics, notably in the realm of double-stranded RNA (dsRNA) viruses. Innovations like Full-Length Amplification of cDNAs (FLAC) have emerged, allowing for the swift generation of full-length cDNA copies of dsRNA genes. This breakthrough facilitates high-throughput sequencing and molecular epidemiology studies, empowering researchers to swiftly generate sequence data from numerous virus isolates. Consequently, these pioneering methodologies enhance the surveillance and management of viral diseases.
FAQ:
Q: What is the purpose of cDNA synthesis in RNA sequencing?
A: Although it is feasible to directly sequence RNA molecules, a significant percentage of RNA-Seq studies utilize instruments designed predominantly for DNA sequencing. This preference is primarily attributable to the advanced technical proficiency of commercially available DNA-based sequencing instruments. Consequently, the creation of a cDNA library from RNA constitutes a prerequisite step in the RNA-Seq procedure.
Q: How much RNA should I use for cDNA synthesis?
A: The requisite amount of RNA for successful cDNA synthesis hinges on two key factors: the sensitivity of the reverse transcriptase and the abundance of the target sequence. Most established protocols propose the use of RNA quantities ranging from 0.1 to 1 µg, although in certain cases, this amount can vary between 100 ng and 20 µg.
Consider, also, the RNA's quality and the transcript of interest's copy number, as they contribute significantly to the Ct value. A prime example is that a high-quality RNA sample of only 100 ng might suffice for a gene that undergoes extensive transcription.
Interpreting the appropriate use of RNA is crucial since an excessive amount can lead to counterintuitive results. It is noteworthy that the transition rate of RNA to cDNA exhibits considerable variability in practical scenarios, with the reverse transcription (RT) efficiency frequently falling below 100%. The factors most commonly linked to suboptimal RT efficiency rates include low-quality primer design, inappropriate reagent concentrations, or unfavorable reaction conditions.