
 Sample Submission Guidelines
Sample Submission Guidelines
How to Detect Histone Modifications by Sequencing?
What Are Histone Modifications?
Histone modifications are intricate chemical alterations that play a pivotal role in the regulation of genetic information stored in DNA. In the genetic code, the sequence of nucleotide bases, adenine (A), cytosine (C), thymine (T), and guanine (G), is fundamental. Surrounding DNA, there are four core histone proteins - H2A, H2B, H3, and H4 - which collectively form a bead-like structure. This assembly, known as a nucleosome, serves as the fundamental building block of chromatin.
Each nucleosome comprises two sets of these core histones creating an octameric structure. The DNA strand wraps around this octamer, spanning approximately 145 to 147 base pairs.
What distinguishes nucleosomes from smooth, uniform beads are the "tails" extending from H2A, H2B, H3, and H4 histones. These tails are comprised of specific sequences of amino acids, and they are subject to post-translational modifications (PTMs). These modifications encompass a range of chemical changes, including methylation, acetylation, phosphorylation, ubiquitination, and more.
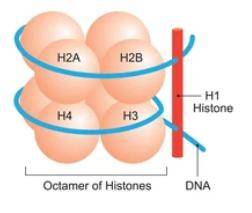 Histone structure.
Histone structure.
Histone modifications exert a profound influence on critical DNA-dependent processes such as chromosome compaction, nucleosome dynamics, and transcriptional regulation. By affecting the level of chromatin compaction and accessibility, these modifications have a significant impact on gene expression. Ultimately, they play a pivotal role in shaping all aspects of biological physiology and developmental processes. In the realm of epigenetics, histone modifications are among the most important mechanisms for regulating gene expression in eukaryotic organisms.
Among these mechanisms, histone acetylation and methylation have undergone extensive research and are recognized as two pivotal and pervasive epigenetic regulatory processes in gene expression.
Histone Methylation
The most thoroughly examined histone modifications encompass methylation and acetylation. Histone methylation alterations are orchestrated by histone methyltransferases (HMTs) and histone demethylases (HDMs). Typically, histone methylation occurs at arginine and lysine residues of H3 and H4. These residues can be mono-, di-, or trimethylated, with lysine also undergoing trimethylation.
Histone arginine methylation augments transcription, while lysine methylation at various positions yields diverse effects, some promoting gene expression and others inhibiting it. The extent of methylation also imparts distinct functions, such as monomethylation and trimethylation of histone H3K4. H3K4me1 typically designates transcriptional enhancers, while H3K4me3 marks gene promoters.
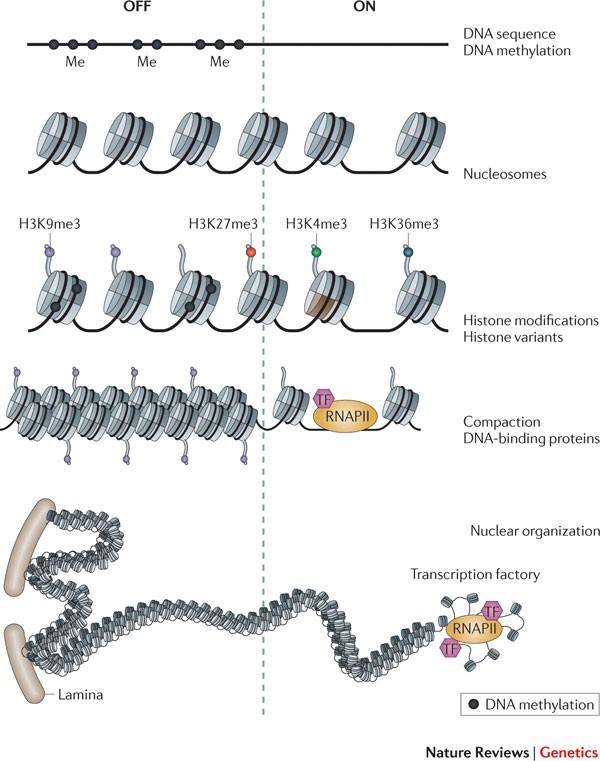 Histone methylation. (Zhou et al., 2011)
Histone methylation. (Zhou et al., 2011)
Histone Acetylation
Histone acetylation is governed by histone acetyltransferases (HATs) and histone deacetylases (HDACs). This modification predominantly occurs on lysines and stimulates the transcription of gene transcripts. The underlying principle is that acetylation modifications neutralize the positive charge of histones, which are attracted to the negatively charged DNA. Greater acetylation leads to reduced histone-DNA binding, granting easier access to DNA for transcription factors and RNA polymerases. For instance, H3K27ac and H3K9ac are frequently associated with enhancer and promoter activity.
Novel Histone Modifications
Apart from the conventional histone modifications mentioned earlier, numerous novel modifications have emerged in recent years. These include lactylation, propionylation, butyrylation, 2-hydroxyisobutyrylation, succinylation, malonylation, glutaralylation, crotonylation, and β-hydroxybutyrylation.
In 2019, Nature published a report titled "Metabolic Regulation of Gene Expression by Histone Lactylation," unveiling the metabolic control of gene expression through histone lactylation. This study not only reintroduced the concept that lactate is more than just a metabolic byproduct but also emphasized its influence on the body's epigenetic modifications. The report brought lactylation, a novel epigenetic modification, to the forefront of scientific attention.
Histone Modification Research Methods: ChIP-seq
ChIP-seq, an acronym for Chromatin Immunoprecipitation followed by Next-Generation Sequencing (NGS), stands as a powerful technique at the forefront of epigenetics research. It seamlessly integrates the strengths of ChIP, which isolates target histone modifications, with the precision of second-generation sequencing. By employing antibodies tailored to specific histone modifications, this method allows for the selective immunoprecipitation of histone-modified DNA fragments. Subsequently, these DNA fragments are fragmented and sequenced, thereby unveiling the precise locations and relative abundances of histone modifications across the entire genome. ChIP-seq, indeed, has evolved into the gold standard for comprehensively elucidating the distribution of histone modifications in the genome.
Histone modifications and DNA methylation, as pivotal epigenetic marks, exert profound influence over chromatin architecture and functionality. They are instrumental in regulating a multitude of essential biological processes, such as gene expression, DNA replication, and repair. The intricate interplay between these two epigenetic mechanisms is vital for maintaining physiological equilibrium and is of critical importance in various disease states.
In contrast to earlier methodologies that necessitated separate experimental approaches for the detection of histone modifications and DNA methylation, ChIP-seq technology, for instance, facilitates the targeted investigation of histone modifications. Complementary techniques like BS-seq are employed for the precise detection of DNA methylation patterns. Importantly, this innovative approach not only enhances research efficiency but also allows for a more comprehensive analysis of epigenetic marks. It is essential to underscore that traditional methods not only entail substantial sample requirements but also lack the capability to simultaneously assess multiple epigenetic marks.
Nanopore Sequencing
The relentless advancement of Nanopore sequencing technology has ushered in a new era of genomics research. Notably, its extended read lengths and capacity for direct sequencing have greatly enhanced the precision of histone modification identification. More significantly, nanopore sequencing technology has the unique capability to concurrently discern DNA methylation and histone modifications. It allows for the probing of the inherent interplay between these epigenetic mechanisms while simultaneously unveiling the spatial arrangement of these marks across the genome, even at low sequencing depths.
Nanopore sequencing holds distinct advantages in terms of its simplicity, heightened sensitivity, and reliability:
- Detection of Histone Modifications and DNA Methylation: This groundbreaking technology enables the concurrent detection of both histone modifications and DNA methylation on a single DNA molecule. It effectively eradicates the need for separate experimental methods to study these two distinct epigenetic markers.
- Extended Read Lengths: Nanopore sequencing delivers extended read lengths, which substantially enhance the precision and sensitivity of detection, providing a more comprehensive view of epigenetic landscapes.
- Streamlined Workflow: The methodology boasts a streamlined experimental procedure, condensing the entire process from cell harvesting to library preparation into a single day.
- Low-Coverage Exploration: Notably, nanopore sequencing allows for simultaneous profiling of DNA methylation and histone modifications even at low sequencing depths. This is instrumental in delving into the intrinsic connections between these two critical epigenetic elements.
Reference:
- Zhou, Vicky W., Alon Goren, and Bradley E. Bernstein. "Charting histone modifications and the functional organization of mammalian genomes." Nature Reviews Genetics 12.1 (2011): 7-18.


