
 Sample Submission Guidelines
Sample Submission Guidelines
Overview of Hi-C Sequencing
What is Hi-C Sequencing?
Genome research encompasses various dimensions, primarily categorized into 1D, 2D, and 3D representations. In the 1D realm, researchers utilize linear mapping techniques to study genomic sequences. Moving into the 2D dimension, they delve into network analysis, particularly focusing on scale-free networks. Finally, the 3D dimension examines the structural and dynamic aspects of the genome.
Hi-C technology stands out as a powerful method for investigating the 3D structure of genomes. Derived from the fusion of High-Throughput Sequencing (HTS) and Chromosome Conformation Capture (3C), Hi-C offers insights into the spatial organization of chromatin within the nucleus.
Chromosome Conformation Capture (3C) involves a series of steps: fixation of nuclear chromatin, digestion of chromatin-protein cross-links, ligation of digests, release of bound proteins, and PCR analysis to detect interactions between DNA fragments. This method assumes that physically interacting DNA fragments exhibit higher linkage frequencies, which are identified through locus-specific PCR.
Hi-C takes this a step further by constructing chromosome-level assemblies of fragmented genomic sequences and determining their order and orientation on the chromosome. Additionally, Hi-C can be integrated with other omics data, such as RNA-Seq and ChIP-Seq, to elucidate gene regulatory and epigenetic networks underlying organismal traits.
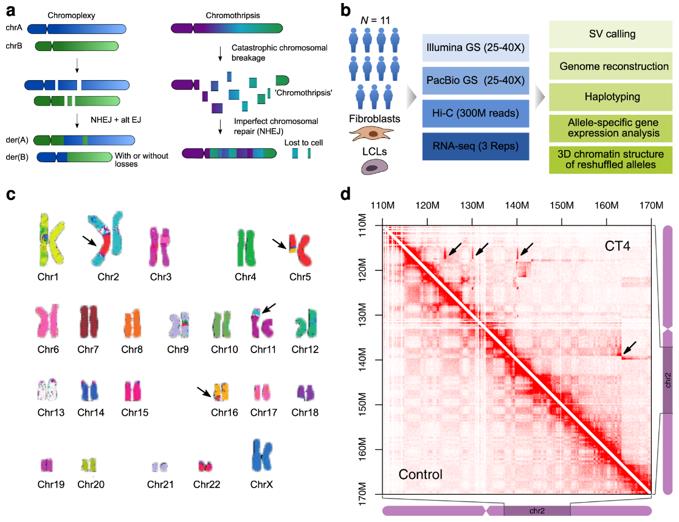 Forms of complex genomic rearrangements: Chromoplexy is characterized by the exchange of larger fragments between chromosomes. (Schöpflin et al., 2022)
Forms of complex genomic rearrangements: Chromoplexy is characterized by the exchange of larger fragments between chromosomes. (Schöpflin et al., 2022)
Advantages of Hi-C Chromosome Construction
- Individual-centric Approach: Unlike traditional methods that often require constructing populations, Hi-C chromosome construction can be effectively executed by a single individual. This streamlines the process, reducing the need for extensive population-based studies.
- Enhanced Localization Efficiency: Hi-C chromosome construction boasts higher localization efficiency compared to many conventional techniques. This means that it can precisely pinpoint the spatial organization of genomic sequences, providing detailed insights into chromatin interactions and structural arrangements.
- Error Correction Capabilities: One notable advantage of Hi-C chromosome construction is its capacity for error correction during the assembly of the genome. This feature allows researchers to refine and improve the accuracy of the assembled genome, ensuring reliable results for downstream analyses and interpretations.
Workflow of Hi-C Sequencing Technology
- Cell Cross-linking
Cells undergo preparation and fixation through cross-linking with either formaldehyde or paraformaldehyde. This process preserves intracellular protein-DNA and DNA-DNA interactions, thus maintaining the 3D structure within the cell. For live samples, a typical treatment involves 1-3% formaldehyde for 10-30 minutes at room temperature. However, it's crucial to note that this step can hinder the efficiency of DNA sequence digestion by restriction endonucleases and requires precise control.
- Endonuclease Digestion
DNA is enzymatically cleaved using restriction endonucleases, generating sticky ends on both sides of the crosslinks. The size of the resulting fragments impacts sequencing resolution. Generally, two enzymes are available for selection: a 6 bp restriction endonuclease or a 4 bp restriction endonuclease. Enzymes such as EcoR1 or HindIII are utilized to cut the genome approximately every 4000 bp, resulting in around 1 million fragments within the human genome.
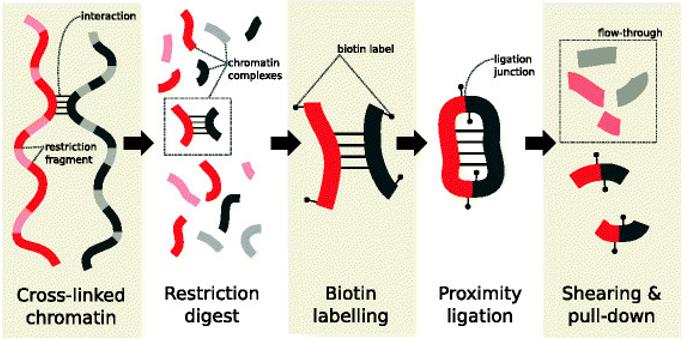 Main steps in the Hi-C protocol prior to sequencing. (Lun et al., 2015)
Main steps in the Hi-C protocol prior to sequencing. (Lun et al., 2015)
- End Repair
The fragmented DNA possesses either flat or sticky ends, which undergo repair to create blunt ends. During this process, biotin-labeled bases are introduced to facilitate subsequent DNA purification and capture.
- Cyclization
End-repaired DNA fragments are looped between DNA segments containing interactions using T4 DNA ligase. Subsequently, proteins connecting the DNA fragments are digested to isolate the cross-linked fragments.
- DNA Purification and Capture
The DNA is de-crosslinked, purified, and fragmented into 300 bp - 700 bp fragments. Fragments containing interactions are then captured for library construction using strand-affinity magnetic beads. Ultrasound or similar methods are employed to further break down the fragments.
- Sequencing
Biotin-containing fragments are captured using magnetic beads, libraries are constructed, and sequencing is carried out.
Data Analysis for Hi-C Sequencing
The data analysis process for Hi-C sequencing entails six critical steps:
- Pre-Raw Reads Filtering: This initial step involves filtering raw reads to remove low-quality or erroneous sequences, similar to standard second-generation sequencing data processing procedures.
- Sequence Comparison: Utilizing pair-end sequencing mode is recommended for Hi-C data analysis, facilitating accurate comparison of sequences.
- Positioning of Cleavage Sites: After determining the physical location of read pairs in the genome, the next step involves identifying the nearest restriction enzyme cleavage site corresponding to each read pair. This is achieved by considering the insertion fragment size restriction. The position of the enzymatically cleaved segment provides an approximate location where DNA interactions occurred.
- Screening for Valid Comparison Fragments: Valid comparison fragments are identified as paired reads located at opposite ends of the cleavage site and mapped in opposite directions, ensuring accurate representation of chromosomal interactions.
- Integration of DNA Fragment Interaction Intensities: The intensities of DNA fragment interactions are integrated, providing insights into the strength and frequency of interactions between genomic loci.
- Normalization of DNA Fragment Interaction Matrix: Normalization of the DNA fragment interaction matrix is performed to ensure unbiased comparisons and accurate interpretation of interaction data.
Hi-C Assembly
Hi-C assembly is typically conducted using software such as LACHESIS, which segments, sequences, and orients the genome based on the support provided by valid read pairs. This process involves manual mapping and verification of the genome to obtain a final chromosome-level assembly.
Valid read pairs yield signals on the map, with the strength of the signal directly proportional to the spatial and sequence distance between contigs. This information allows for error correction, including the identification and correction of misassembled contigs, adjustment of contig orientations, and determination of contig placement within chromosomes through clustering.
Ultimately, a refined chromosome-level genome assembly is obtained, with any remaining discrepancies fine-tuned manually to ensure accuracy. The manual adjustment process aims to achieve a clear diagonal signal, indicative of a well-assembled genome.
Applications of Hi-C Technology
In recent years, Hi-C technology has played a pivotal role in advancing genome assembly and understanding the three-dimensional (3D) structure of genomes across various organisms including humans, goats, mosquitoes, yeast, barley, and wheat. The successful assembly of chromosome-level genomes in these species underscores the reliability and versatility of Hi-C-assisted genome assembly technology.
Hi-C technology unveils the intricate three-dimensional structure of genomes, elucidating the hierarchical organization of chromatin from compartments (A/B Compartments) to topology-associated structural domains (TADs), and further to loops. This comprehensive understanding is crucial for studying spatial interactions among DNA sequences, constructing high-resolution chromosome 3D structures, deciphering gene regulation mechanisms, and facilitating the construction of trans-chromosomal genomes and chromosome-spanning haplotypes. Notably, genome 3D structures have been successfully reconstructed in various organisms including humans, Drosophila, yeast, Arabidopsis thaliana, rice, and cotton species. Comparative analysis of genome 3D structures across different samples has also been accomplished, shedding light on evolutionary and functional insights.
- Whole Genome Hi-C Maps: This includes comprehensive analysis of cis/trans interactions within the genome.
- Compartment A/B Identification and Analysis: Hi-C facilitates the identification and analysis of genomic compartments, complemented by joint analyses with Chip-seq and RNA-seq data.
- Gene/Repeat Sequence Interactions Analysis: Hi-C enables the investigation of interactions between genes and repetitive sequences, often integrated with RNA-seq data for comprehensive analysis.
- TAD Identification and Analysis: Hi-C technology aids in the identification and analysis of Topologically Associated Domains (TADs), often coupled with Chip-seq and RNA-seq analyses.
- Genome-Wide Loop Modeling: This aspect requires complementary data such as DNase-seq and is often analyzed jointly with Chip-seq and RNA-seq data.
- Differential Analysis of 3D Structures: Hi-C allows for differential analysis of 3D structures among multiple samples, including differential analysis of compartment A/B, TADs, and loops.
References:
- Schöpflin, Robert, et al. "Integration of Hi-C with short and long-read genome sequencing reveals the structure of germline rearranged genomes." Nature Communications 13.1 (2022): 6470.
- Lun, Aaron TL, and Gordon K. Smyth. "diffHic: a Bioconductor package to detect differential genomic interactions in Hi-C data." BMC bioinformatics 16 (2015): 1-11.


