Sample Submission Guidelines
Sample Submission Guidelines
BSA-seq Technology Workflow
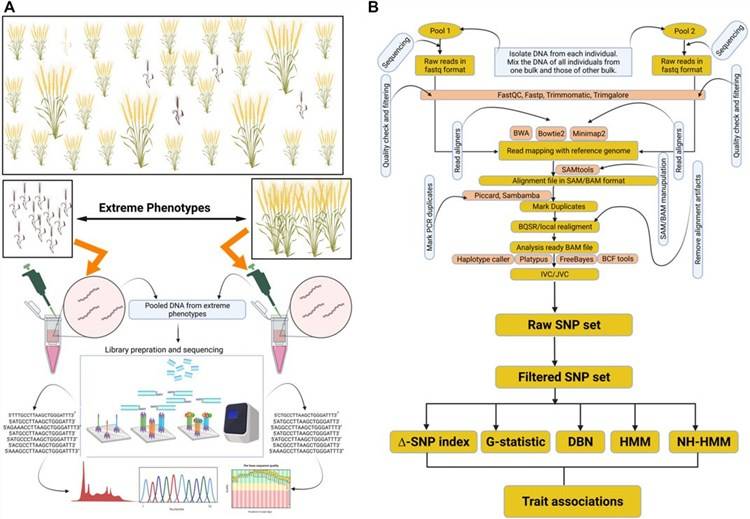
Bulked segregant analysis (BSA) is a method first applied in lettuce by R. W. MICHELMORE in 1991 for the rapid localization of genes controlling specific traits. The method involves selecting 12-14 individual plants with extreme phenotypes from an F2 population, pooling their DNA equally to create two DNA pools. Then, polymorphic markers are screened between the parents and the two pools. If a marker displays consistent polymorphism between the parents and the pools, it is likely linked to the trait. Genotypic analysis of these selected polymorphic markers in the F2 population allows for the localization of the target gene, eliminating the need for genotypic analysis of every marker in the population.
The principle behind this method is that markers linked to the trait will exhibit polymorphism between the two pools, while markers not linked or distantly linked to the target gene will show random heterozygosity between the pools. BSA is a fast way to obtain molecular markers linked to the trait, typically used for locating qualitative trait genes or quantitative trait loci (QTL) controlling traits with a small number (2-3) of major effects.
DNA Sample Extraction And Detection
First, DNA is extracted using a DNA extraction kit following a standard procedure. DNA from the two extreme phenotype parents can be directly extracted and quality-checked. In contrast, offspring samples need to undergo an additional step after DNA extraction and quality-checking, involving equimolar mixing to create pooled samples (each pool should ideally contain a minimum of 20 or more samples).
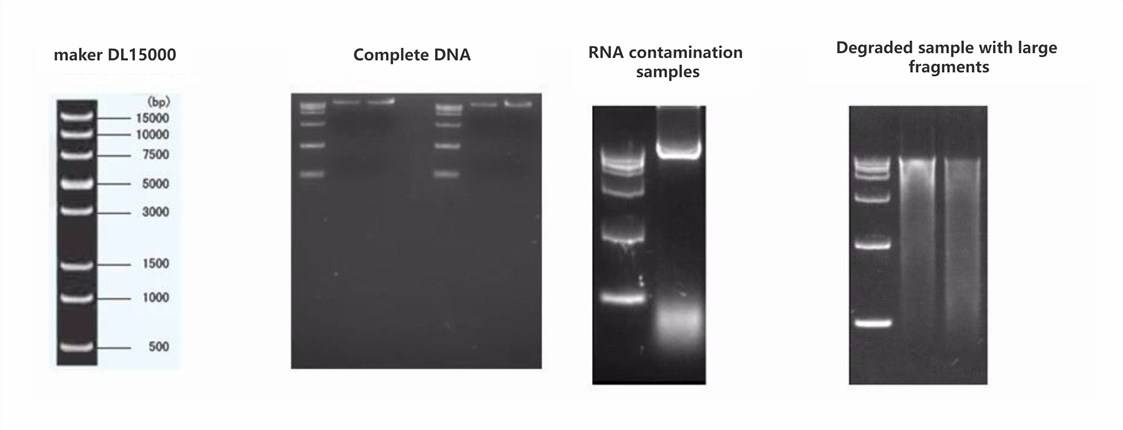
DNA sample detection involves three main methods:
- Agarose gel electrophoresis to analyze the purity and integrity of DNA.
- Nanodrop to assess DNA purity (OD260/280 ratio).
- Qubit for precise quantification of DNA concentration.
Sample DNA requirements: For each library preparation, 2 μg of sample is needed, with at least two preparations provided. The sample concentration should be >20 ng/μl, the OD260/280 ratio should range between 1.8 and 2.0 with no visible contamination, and the genomic DNA should be intact without degradation. In gel electrophoresis, the DNA's main band should be larger than 23 kb.
Sample selection: For plant samples, it is recommended to choose dark-cultured yellowing seedlings or tender seedlings. For animal samples, select tissues with low fat content, such as muscle or blood, for sampling.
Library Construction
DNA samples that have passed quality control are fragmented into 350 bp fragments using a fragmenting reagent kit for library construction. The DNA fragments undergo various steps, including end repair, polyA tail addition, sequencing adapter ligation, purification, PCR amplification, to complete the entire library preparation process. After library construction is completed, an initial quantification is performed. Subsequently, the insert fragment length of the library is checked, and once the length matches the expected size, quantitative PCR (qPCR) is used to accurately determine the effective concentration of the library to ensure its quality. Once the library passes these quality checks, it proceeds to the next stage of sequencing.
The specific workflow for library construction and sequencing is illustrated in the following diagram:
 Aasim Majeed et al,. Front. Genet., 08 August 2022
Aasim Majeed et al,. Front. Genet., 08 August 2022
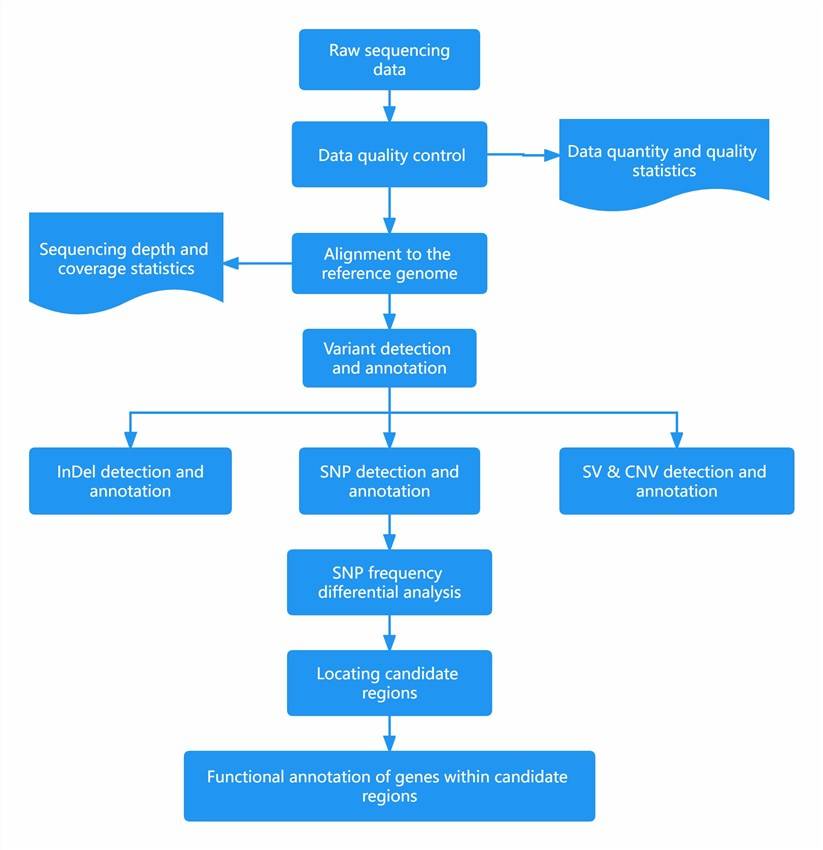
Bioinformation analysis process
Once the quality control process is complete, the library construction and sequencing workflow proceeds as follows:
(1) Data Quality Control:
After obtaining the raw sequencing data (Sequenced Reads), adaptor sequences and sequences like polyN and polyA are filtered out to obtain clean data.
(2) Alignment to the Reference Genome:
The filtered valid reads are aligned to the reference genome using BWA (Burrows-Wheeler Aligner). The SAMtools software is then used to sort the alignment results, followed by using Picard to mark duplicate reads. Marking duplicate reads involves labeling multiple identical DNA fragments amplified through PCR. Labeled reads are not used in subsequent analyses to prevent false positives in variant detection.
(3) SNP and InDel Detection:
After marking duplicate sequences, it is necessary to realign InDels based on the CIGAR (Compact Idiosyncratic Gapped Alignment Report) values provided in the BWA alignment results. BWA tolerates mismatches (mismatches and InDels) near or within continuous runs of homopolymer nucleotides (e.g., consecutive T's or A's). This can lead to errors in variant calling. Therefore, using the GATK (Genome Analysis Toolkit) software's InDel Realignment module is essential to minimize alignment errors near InDels (Insertion-Deletion).
It's important to note that the quality of base calls (Quality scores) is crucial for analysis. However, sequencing instruments introduce systematic biases that can significantly impact downstream analysis. Before base quality score recalibration, for example, base calls with quality scores above Q25 are retained. In reality, bases with a quality score of Q25 have a 1% error rate. Thus, having a quality score of Q20 may affect the credibility of subsequent variant detection. Errors in base calls at the ends of reads are often higher than at the beginning. Additionally, the quality of AC bases is typically lower than that of TG bases. Therefore, GATK's Base Recalibration is employed to correct base quality scores, ensuring more consistent and reliable sequencing quality. Note: In cases of multiple sequencing runs for the same sample or multiple samples in different lanes, base recalibration should be performed separately for each lane to ensure accuracy and effectiveness.
Following these preliminary steps, the next phase involves the detection of mutation sites (variant calling). Distinguishing true genetic variants from potential sequencing errors (random machine noise) is a critical challenge in this process. GATK offers two methods: the model-independent UnifiedGenotyper, which doesn't consider the influence of adjacent bases, and the local de-novo model-based HaplotypeCaller. The HaplotypeCaller constructs a DeBruijn graph and employs the PairHMM model for single haplotype prediction and mutation site reliability assessment, resulting in more accurate variant detection.
Currently, the UnifiedGenotyper tool, in combination with previous InDel realignment and Base recalibration, allows for precise SNP detection.
(4) Structural Variation (SV) Detection:
Structural variations on the genome are analyzed using the Lumpy algorithm.
(5) Copy Number Variation (CNV) Detection:
Analysis of Copy Number Variations on the genome is performed using the Control-Freec algorithm, which can predict copy numbers for each changing region.
(6) Annotation:
Biological information annotation for mutation sites in coding regions is vital, as these regions are critical for disease occurrence and trait changes. SnpEff and Annovar software are used for structural annotation of mutation sites.
(7) Candidate Region Localization:
Based on the detected SNP sites, the SNP-index of pooled samples is calculated, along with the frequency difference between the two extreme trait pools. Significant difference regions are selected for candidate region localization.
(8) Functional Annotation of Genes Within Candidate Regions:
For candidate genes within the candidate region, GO (Gene Ontology) and KEGG (Kyoto Encyclopedia of Genes and Genomes) functional annotation is performed.
(9) Distribution and Type Statistics of SNP and InDel Within Candidate Regions:
Analysis is carried out to understand the relationship between SNP and InDel variations within candidate regions and annotated genes. This includes analyzing regions where non-synonymous substitutions or variants causing premature stop codons occur, as well as regions where variation occurs in regulatory regions (promoter regions).