
 Sample Submission Guidelines
Sample Submission Guidelines
Bioinformatic Analysis of Chloroplast Genome
Chloroplast Genome
Chloroplasts are a ubiquitous feature in plants, housing the chloroplast genome, a double-stranded circular DNA molecule. A single plant contains multiple chloroplasts, each harboring 12 cpDNA molecules.
The typical size of a plant's chloroplast genome ranges from 150 to 160 kb, whereas algae tend to have slightly smaller genomes of around 80 to 100 kb. These genomes are organized into four distinct regions: the Large Single Copy (LSC), the Small Single Copy (SSC), and two Inverted Repeat (IR) regions situated between the LSC and SSC. As high-throughput sequencing technology advances at an unprecedented rate, the utilization of chloroplasts as a means to investigate the origin, structure, and evolution of organelles is garnering increasing attention and interest.
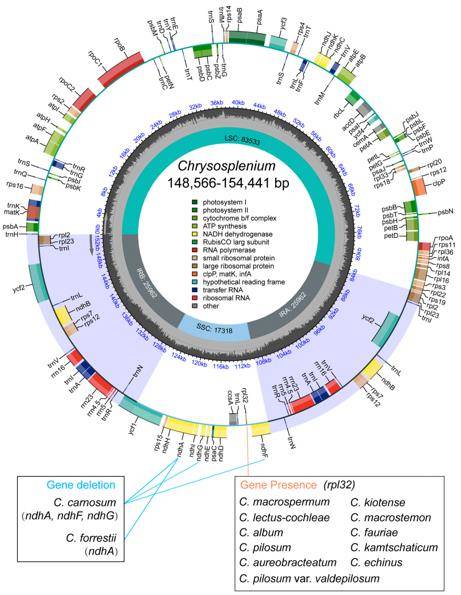 Representative chloroplast genome map of Chrysosplenium. (Yang et al., 2023)
Representative chloroplast genome map of Chrysosplenium. (Yang et al., 2023)
Covariance Analysis in Genetics
Covariance, in the context of genetics, refers to the phenomenon of gene linkage. It occurs when homologous genes are arranged in the same order on the chromosomes of different species. The extent of covariance between two species serves as a valuable metric for gauging their evolutionary divergence and assessing their genetic relatedness.
Conducting a covariance analysis involves studying specific, localized blocks of covariance within genomes. This analysis can shed light on various evolutionary events, including similarities, rearrangements, inversions, and other genetic alterations that have taken place during the course of species evolution.
Phylogenetic Tree Analysis
A phylogenetic tree, also known as a phylogeny, is a branching diagram that visually represents the relationships between species, allowing us to understand their evolutionary history. Phylogenetic tree analysis serves several key purposes, including identifying the evolutionary connections between species, uncovering the links between ancestral and descendant sequences, and estimating the time of divergence among a group of species that share a common ancestor.
Organelle genomes, due to their high conservation, are frequently employed to construct phylogenetic trees for the classification and assessment of the evolutionary status of both plants and animals. Two methods are available for constructing organelle phylogenetic trees:
- Population SNP Matrix-Based Tree Construction: This approach involves the generation of an evolutionary tree using the population SNP matrix derived from samples and reference genomes. For each sample, all Single Nucleotide Polymorphisms (SNPs) are aligned in the same order, producing sequences of the same length in fasta format, including a reference sequence. These aligned sequences are then employed as input data for constructing the phylogenetic tree.
- Core Gene-Based Tree Construction: In this method, phylogenetic trees are constructed based on core genes that are present as single-copy genes within the organelle genome. The multiple sequences of these core genes are aligned using MUSCLE v3.8.31 software, and the resulting alignment data is used to build the evolutionary tree, revealing the genetic relationships among species.
Detection of Structural Variations in Chloroplast Genomes
The process of detecting structural variations in organelle genomes primarily encompasses three types: Single Nucleotide Polymorphisms (SNP), Insertions/Deletions (InDel), and Structural Variations (SV). Comparing organelle genomes of closely related species to reference genomes is a crucial step in understanding and analyzing the variations present within individuals or populations.
- SNP (Single Nucleotide Polymorphism)
SNPs are variations in DNA sequences resulting from a single-nucleotide change. These changes can occur within coding genes or non-coding sequences. SNPs within coding regions, known as coding SNPs (cSNPs), are particularly important as they have the potential to impact an individual's functional attributes.
- InDel (Insertion/Deletion)
InDel serves as a collective term for the insertion and deletion of DNA sequences. In a more specific context, narrow InDel refers to relatively short insertions or deletions, typically ranging from 1 to 10 base pairs. In the coding regions of the genome, InDel events can lead to frame-shift mutations, alterations in amino acid sequences, and even the formation of pseudogenes. The focus here is on the analysis of these narrow InDel variations.
- Structural Variation (SV)
Structural Variations encompass a range of genomic alterations such as deletions, insertions, duplications, inversions, and ectopic relocations of DNA fragments within the genome. To identify SVs, the MUMmer software is employed to compare the target organelle genome with a reference genome. Subsequently, LASTZ is utilized to perform region-to-region comparisons, thereby pinpointing SVs in the organelle genome through the analysis of the comparison results.
Analysis of Common and Specific Genes
Within a set of genetic samples, genes that share homology across all samples are referred to as 'core genes.' Conversely, genes that are not common after the core genes are removed are categorized as 'dispensable genes.' 'Specific genes' denote genes that are unique to a particular sample. These shared and specific genes often correspond to the common attributes and distinctive characteristics of the respective samples. They serve as a fundamental basis for exploring functional disparities between samples.
Codon Preference Analysis
Codon preference, also known as codon usage bias, quantifies the relative likelihood of a specific codon appearing among synonymous codons that encode the same amino acid. The value for codon preference is typically determined through the calculation of Relative Synonymous Codon Usage (RSCU). The study of codon usage patterns holds significant importance, as it provides insights into the evolutionary pressures on species and plays a crucial role in advancing genetic investigations.
Analysis of Simple Sequence Repeats (SSR)
Simple Sequence Repeats (SSR), also known as microsatellites (MS), are DNA fragments characterized by the repetition of short sequences comprising 1-6 nucleotides. These SSRs are abundant, highly polymorphic, evenly distributed throughout the genome, co-dominant, and relatively straightforward to detect. As a result, they have found extensive applications as second-generation molecular markers in various genetic research areas, including genetic mapping, the localization of target genes, investigations into genetic diversity, the identification of germplasm resources, and molecular-assisted breeding. SSRs are a cornerstone in genetic research, contributing significantly to genetic map construction and resource identification.
Reference:
- Yang, Tiange, et al. "A Comprehensive Analysis of Chloroplast Genome Provides New Insights into the Evolution of the Genus Chrysosplenium." International Journal of Molecular Sciences 24.19 (2023): 14735.


