
 Sample Submission Guidelines
Sample Submission Guidelines
miRNA Sequencing Data Analysis Service
Introduction of miRNA sequencing Data Analysis
With the advancement of high-throughput sequencing technologies, miRNA-seq has become a powerful tool for profiling miRNA expression in various samples. The analysis of miRNA sequencing data is crucial for advancing our understanding of gene regulation, disease mechanisms, and personalized medicine. It allows for the discovery of regulatory miRNAs involved in important biological processes and the identification of miRNAs as potential biomarkers for disease diagnosis, prognosis, and treatment response. Furthermore, miRNA sequencing data analysis helps unravel complex miRNA-mediated regulatory networks and aids in the development of personalized medicine approaches. Integrating miRNA sequencing data with other omics data enhances our understanding of biological processes and allows for the development of targeted therapies.
miRNA Sequencing Data Analysis Service
CD Genomics' team of professional bioinformatics experts, with extensive project experience, provides miRNA sequencing data analysis services, including standard analysis, advanced analysis, and customized analysis to meet your specific research needs.
miRNA-Seq Data Analysis Workflow
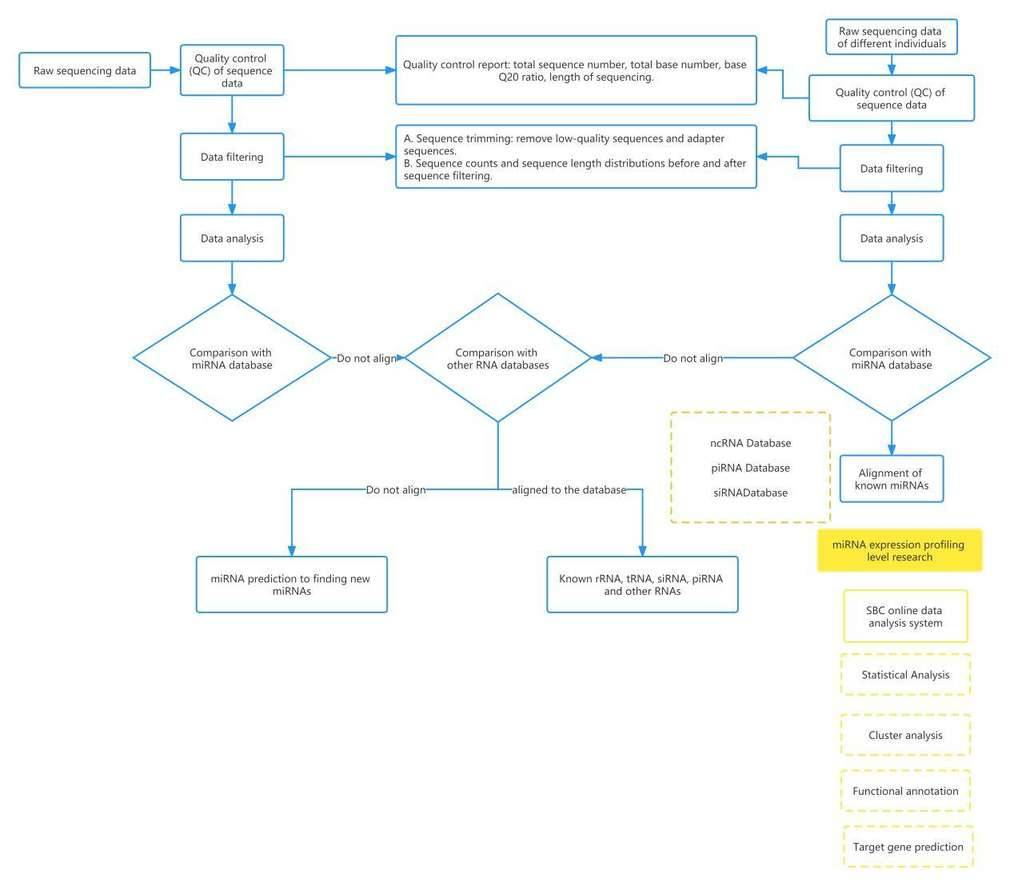
Data Analysis Content
1. MicroRNA length distribution statistics
Apply fastx (fastx_toolkit-0.0.13.2) to preprocess the original reads, remove adapter sequences and low-quality sequences (including ambiguous base N, sequences with base quality less than 10 and length less than 18nt), and finally give the statistics of the processing results Table and length distribution diagram.
2. MicroRNA Alignment Annotation Statistics
The sequence obtained by sequencing was compared with the Sanger miRBase database, known rRNA, tRNA, repeat region, RefSeq database, and other non-coding databases such as ncRNA, piRNA, Rfam database, and annotated the known microRNA.
3. Annotation of Other Small RNAs
The sequenced reads are aligned to the corresponding genomic databases for the species, and the sources of annotated reads are classified and counted. This process helps identify and quantify known microRNAs and other different types of small RNA molecules.
4. Prediction of Novel microRNAs
The characteristic hairpin structure of microRNA precursors can be used to predict novel microRNAs. For sequences that are not annotated in the alignment results, they are compared to the whole genome sequence of the species. By analyzing the folding models, if a sequence is located on a stem-loop structure, it is tentatively identified as a candidate novel microRNA.
For the predicted novel microRNAs, their chromosomal locations, start and end positions, strand orientation, as well as numerical values such as count, length, GC content, and minimum free energy are recorded and listed.
Additionally, for the novel microRNAs, the secondary structure of their precursors and their relationship with mature microRNAs are calculated and presented.
5. Saturation Analysis Charts
Saturation analysis examines whether the detection of microRNAs listed in the miRBase database increases with the increase in sequencing depth. The annotation results are proportionally divided and plotted to observe the trend of annotation results in biological samples, ensuring their biological validity.
6. Selection of microRNA Expression
Differences between Samples DEGseq R package and Perl scripts are used to compare the expression levels of microRNAs between samples based on the customer's grouping criteria (e.g., control group vs. experimental group). In the differential analysis, the TPM (Transcripts per million) is used as the normalized data, calculated as the individual microRNA read count multiplied by 10^6 divided by the total read count.
7. Prediction of microRNA
Target Genes The miranda software is employed to predict potential target sites for microRNA sequences and the corresponding genomic cDNA sequences of the species.
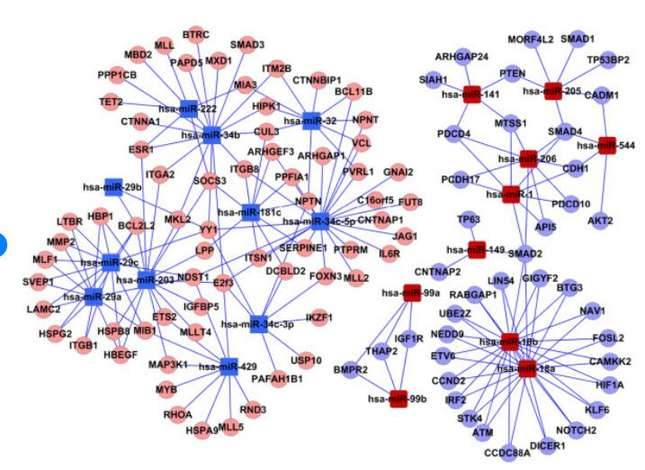 (Zhaohui Luo, et al,. 2012)
(Zhaohui Luo, et al,. 2012)
Alternatively, the TargetScan database can be used to associate microRNAs with their target genes. The results include relevant information on the microRNA and its target genes.
8. GO Functional Enrichment Analysis (Target Genes)
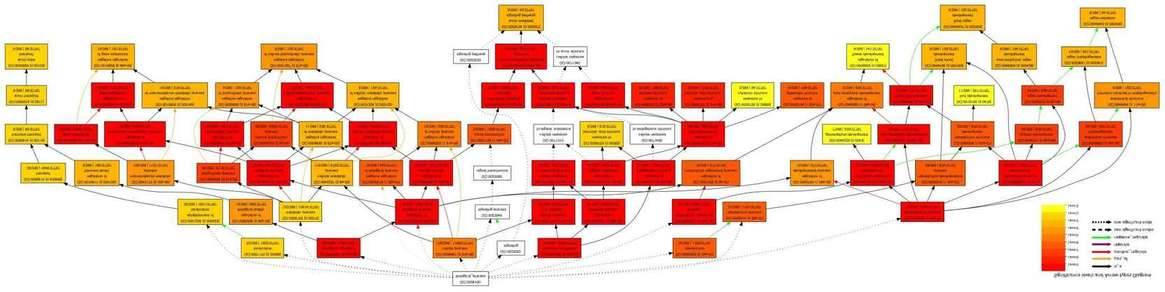 (Yuepeng Song et al,. 2013)
(Yuepeng Song et al,. 2013)
9. Pathway Significance Enrichment Analysis (Target Genes)
Case 1:Sexual dimorphism floral microRNA profiling and target gene expression in andromonoecious poplar (Populus tomentosa). PLoS One. 2013, 8(5):e62681.(IF3.730)
This study reports for the first time the identification of miRNAs associated with flower development and their target genes. Based on gene annotation, all the target genes were classified into four categories.
The first category is flower development-related genes, including Pto-F6, Pto-F11, Pto-F14, Pto-F19, Pto-25, Pto-36, Pto-F51, Pto-F54, and Pto-F66.
The second category is calcium ion transport-related genes: Pto-F28 and Pto-F45 (female flower-specific miRNAs) and Pto-F16 (male flower-specific miRNA).
The third category is hormone regulation-related genes: 10 miRNAs (excluding Pto-F6) that are predominantly expressed in female flowers.
The fourth category is DNA methylation-related genes: three miRNAs.
These findings provide insights into the regulatory roles of miRNAs in flower development, including their involvement in flower organogenesis, hormone signaling, calcium ion transport, and DNA methylation processes.
Case 2:MeCP2 Suppresses Nuclear MicroRNA Processing and Dendritic Growth by Regulating the DGCR8/Drosha Complex p547. Developmental Cell.2014,28, 547–60.(IF:10.366)
Mutations or copy number variations in the human MeCP2 gene lead to developmental neurological disorders, such as Autism. Researchers from the Institute of Neuroscience, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, led by Dr. Zilong Qiu, utilized high-throughput sequencing technology to quantitatively analyze mature small RNAs in the hippocampal region of MeCP2 knockout mice. They discovered that MeCP2 inhibits the production of a large number of small RNAs. This study revealed a novel function of the autism-associated protein MeCP2 in small RNA processing and suggests that this function may be involved in the pathogenic mechanism underlying developmental neurological disorders caused by MeCP2 gene mutations.


