
 Sample Submission Guidelines
Sample Submission Guidelines
Genomic Sequencing of SARS-CoV-2 Accelerates The Identification of Viral Variants And Evolutionary Analysis
Coronaviruses are important pathogens in humans and vertebrates. It can infect the respiratory, gastrointestinal, hepatic and central nervous systems of many wildlife species, including humans, animals, birds, bats and mice.
The potential for transmission of CoVs from animals to humans has been demonstrated since the outbreak of severe acute respiratory syndrome (SARS) in 2002 and Middle East respiratory syndrome (MERS) in 2012. The intermittent emergence and outbreaks of novel coronaviruses remind us that coronaviruses remain a serious global health threat.
With climate and ecological changes and increased human-animal interactions, outbreaks of novel CoVs seem inevitable and effective treatment regimens and vaccines must be developed as soon as possible.
Scientists are continuously working to excavate the genomes of new coronaviruses, trying to understand the origin of 2019-nCoV by probing the genomes of an increasing number of publicly available viruses, as well as to understand how he replicates and how he escapes human defenses and methods of control.
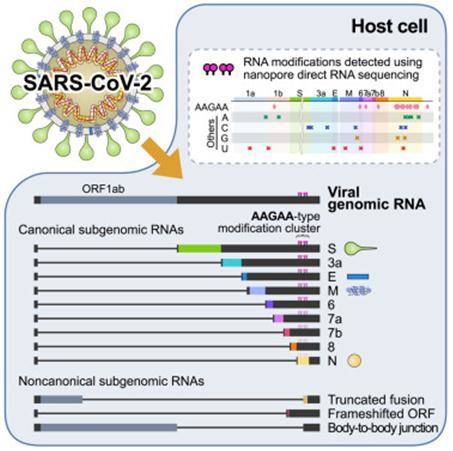
Mapping the genome and subgenomic RNA structure of SARS-CoV-2
The virus is just a piece of bad news wrapped in a protein. For the new coronavirus SARS-CoV-2, which is sweeping the world, this bad news is in the form of a very long RNA carrying the genome. As an RNA virus, SARS-Cov-2 enters the host cell to replicate genomic RNA and produces many smaller RNAs (called subgenomic RNAs). These subgenomic RNAs are used to synthesize the various proteins (spinosyn, capsular proteins, etc.) required for SARS-Cov-2 to begin. Thus, smaller RNAs are good targets for disrupting neo-coronavirus attacks on our immune system.
The researchers combined two complementary sequencing technologies: DNA nanoball sequencing and nanopore direct RNA sequencing to resolve the high-resolution RNA genome structure of SARS-CoV-2 and obtain all SARS-CoV-2 RNAs (transcripts) and all modified RNAs (epitranscriptome). Nanopore direct RNA sequencing allows direct analysis of entire long viral RNAs without fragmentation. Traditional RNA sequencing methods typically require a stepwise process of cutting the RNA and converting it to DNA before reading it. meanwhile, DNA nanosphere sequencing can only read short fragments, but has the advantage of analyzing a large number of sequences with high precision. These two techniques are highly complementary in the analysis of viral RNA. This map will help to understand how the virus replicates and how it escapes the human defense system.
Analysis of SARS-CoV-2 variation and evolution using population genetics
Viruses evolve extremely rapidly, and as the number of infections increases explosively, many different strains of neo-coronaviruses can develop rapidly. It is particularly important to investigate the history of virus genealogy, to typify different strains, and to combine epidemiological and clinical data to reveal the mutation pattern of coronaviruses for epidemic prevention and control.
Using population genetic analysis, the evolutionary history of SARS-CoV-2 populations was deduced from the genomic perspective by analyzing the variation of neo-coronaviruses. The main results of the article are to characterize the genomic variation of new coronaviruses and to infer the evolutionary relationships of global samples.
The haplotype-based phylogenetic analysis provides a powerful approach to understanding the evolution of SARS-CoV-2 in the early stages of transmission when reverse mutations and illegal recombination are rare.
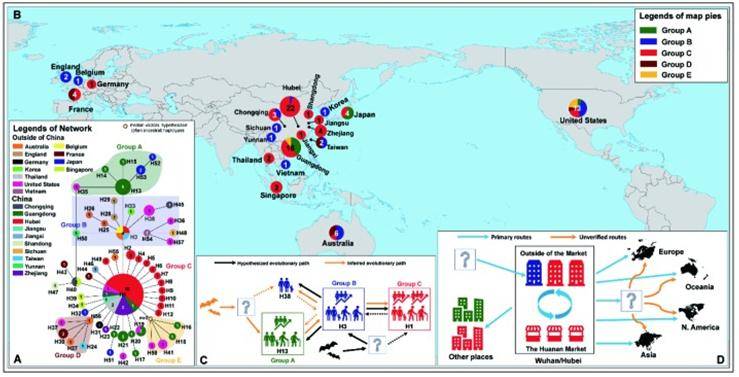 Evolutionary relationship and geographical distribution of 58 haplotypes of SARS-CoV-2 (Yu W B et al., 2020)
Evolutionary relationship and geographical distribution of 58 haplotypes of SARS-CoV-2 (Yu W B et al., 2020)
A total of 120 substitution sites and 119 codons, including 79 nonsynonymous and 40 synonymous substitutions, were identified in the eight coding regions of the SARS-CoV-2 genome. Forty nonsynonymous substitutions may be associated with viral adaptation.
Summary
It will help develop novel antiviral drugs by designing small molecules or interfering RNAs to directly target neo-coronavirus RNAs by revealing the natural genomic structure of coronaviruses in cells and key RNA elements of the viral life cycle
Recommended Services
With years of experience in microbiomics and genomics research, CD Genomics offers viral genome sequencing, metagenome sequencing and population genetics analysis services to help you discover more.
References:
- Kim D, Lee J Y, Yang J S, et al. The architecture of SARS-CoV-2 transcriptome. Cell, 2020, 181(4): 914-921. e10.
- Yu W B, Tang G D, Zhang L, et al. Decoding the evolution and transmissions of the novel pneumonia coronavirus (SARS-CoV-2/HCoV-19) using whole genomic data. Zoological research, 2020, 41(3): 247.


