
 Sample Submission Guidelines
Sample Submission Guidelines
Genetic Variation: Definition, Types, and Variant Calling Workflow
What is Genetic Variation
The genetic makeup of organisms within a population change, which is referred to as genetic variation. Genes are inherited DNA segments that contain the instructions for making proteins. Alternate versions of genes, known as alleles, determine distinct traits that can be passed down from parents to offspring. Natural selection and biological evolution rely heavily on genetic variation. Natural selection does not occur by accident, but genetic variations that arise in a population do.
Transposons are another important term associated with genetic variation. Mutations can be caused by transposons in a variety of ways. A transposon will almost certainly damage a functional gene if it inserts itself into it. To disrupt or alter the activity of a gene, exons, introns, and even DNA flanking the genes (which may contain promoters and enhancers) can all be implanted into.
You may interested in
What are the Types of Genetic Variation
Single base-pair substitution
SNPs (single nucleotide polymorphisms) are nucleic acid substitutions that are also known as single nucleotide polymorphisms (SNPs). (1) transition, which involves the exchange of purine (Adenine/Guanine) or pyrimidine (Cytosine/Thymine) nucleic acids, and (2) transversion, which involves the exchange of purine and pyrimidine nucleic acids.
Insertion or deletion
Insertion or deletion of a single stretch of DNA sequence that can vary in length from two to hundreds of base pairs, also known as 'indel'.
Structural variation
Genetic variation that happens over a larger DNA sequence is commonly referred to as chromosomal variation. Both copy number variation and chromosomal rearrangement events are included in this category of genetic variation.
Copy Number Variation
The phenomenon of copy number variation (CNV) occurs when sections of the genome are repeated and the number of repeats varies between individuals.
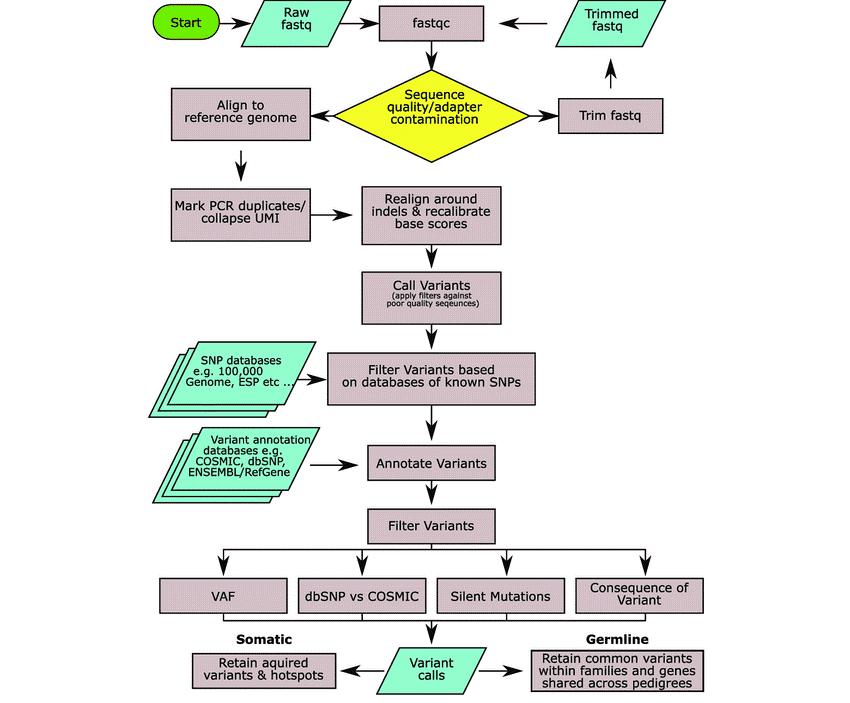 Figure 1. A generalised workflow for calling variants. (Bewicke-Copley, 2019)
Figure 1. A generalised workflow for calling variants. (Bewicke-Copley, 2019)
How dose Variant Calling Work
Variant calling from raw read data is a multistep process that can be done with a wide range of tools and resources. The following are the steps in the procedure:
- To generate FASTQ files, sequence the entire genome or exome.
- Align the sequences to a reference genome to generate BAM or CRAM files.
- Make a VCF file by determining where the aligned reads differ from the reference genome.
Acquirement of raw read data: the FASTQ file configuration
The most popular approach to get raw data from a sequencing machine is through FASTQ files, which are similar to FASTA files and contain sequence information as well as extra information such as sequence quality information.
Quality Control
Raw sequence data obtained from a sequencing service provider, in general, is not immediately ready for variant discovery. Quality control (QC), which comes after data acquisition, is the first and most important phase in the WES/WGS assessment framework. QC is a method for improving raw data by removing any errors that can be detected. By conducting quality control (QC) at the start of the assessment, the chances of encountering contamination, bias, error, or missing data are reduced.
Sequence Alignment
Each read must be aligned to a reference genome to determine its precise location. Because aligning a large number of reads can take days, and a low-accuracy alignment will result in insufficient analyses, reliability and accuracy are critical during this phase. A Sequence Alignment Map (SAM) file is created once the alignment is complete.
Post-Alignment Processing
Post-alignment data processing to construct analysis-ready BAM files is essential in any reads-to-variants framework. This procedure entails cleaning data to remove technical biases, such as identifying duplicates and recalibrating base quality scores.
Short Variant Discovery
After going through data processing steps, the reads are ready for downstream analysis, with variant calling being the most common phase. Variant calling is a method of classifying differences between sequencing reads generated by NGS experiments and a reference genome. Due to the difficulty of variant calling due to alignment and sequencing artifacts, a plethora of variant callers have been developed and are being developed to assist with this difficult task.
Filtration of Variants
After the variant calling stage, raw SNV and indels in the Variant Call Format (VCF) are obtained. After that, either hard filters on the data or a more complex method like GATK's Variant Quality Score Recalibration (VQSR) are employed to filter them.
Variant Annotation
Variable annotation is another crucial process in the WES/WGS assessment framework. The goal of all functional annotation instruments is to annotate data about variant effects/consequences, such as identifying which gene(s)/transcript(s) are influenced, (ii) assessing the influence on protein sequence, and (iii) equating the variant with known genomic annotations, and (iv) finding and complementing known variants in variant databases. The effect of each variant is shown using Sequence Ontology (SO) terms. Qualifiers are frequently used to denote the seriousness and impact of these consequences.
References:
- Bewicke-Copley F, Kumar EA, Palladino G, et al. Applications and analysis of targeted genomic sequencing in cancer studies. Computational and structural biotechnology journal. 2019 Jan 1;17.
- Bedo J, Goudey B, Wazny J, Zhou Z. Information theoretic alignment free variant calling. PeerJ Computer Science. 2016 Jul 25;2.
- Muzzey D, Evans EA, Lieber C. Understanding the basics of NGS: from mechanism to variant calling. Current genetic medicine reports. 2015 Dec;3(4).


