
 Sample Submission Guidelines
Sample Submission Guidelines
Full-length Transcript Sequencing: A Comparison Between PacBio Iso-Seq and Nanopore Direct RNA-Seq
Overview
To properly utilize the underlying genome assemblies, complete and precise gene annotations are important. The production of high-efficiency short-read RNA sequencing (or RNA-seq) innovations has significantly advanced genome research by facilitating both the enhancement of genome annotations and the experiment of organisms that are not yet accessible for reference genomes. Reference-free as well as reference-based transcript assemblies of short reads, even so, are difficult and often do not align gene models that have been experimentally verified. Indeed, the annotation of intricate genomes, such as those of agricultural-relevant plant species, does not yet provide optimal results. The inconsistency of the underlying genome assembly, in relation to imperfect gene model rebuilding, complicates gene annotations.
All the genes displayed in the cell or tissue are represented by the transcriptome. RNA sequencing (RNA-Seq) allows the identification of these genes to be captured. For the experiment of difference in gene expression and the impact of genotype or environment on their expression, creating a reference transcriptome is crucial. By short-read sequencing, most researches generate a reference transcriptome and recreate the transcriptome through the assembly and/or mapping of reads to other accessible reference genomes. For lengthy transcripts, repetitive sequences, and transposable elements, nevertheless, this is complicated.
For intricate polyploid genomes, it is especially challenging. Lately, long-read sequencing (LRS) technology, represented by PacBio Sequencing and Nanopore Sequencing, has become obtainable, and this technology overwhelms these challenges by creating full-length sequence data as a single read sequence, including lengthy transcripts (e.g. those greater than 10 kb) without the need for further assembly. In a few plant research cases, this method has been used and offers additional data on transcript differences, such as alternative splicing and alternative polyadenylation.
Comparison Between PacBio Iso-Seq and Nanopore Direct RNA-Seq
The biggest sequencing potential is provided by Pacific Biosciences (PacBio) Technology (Isoform-Sequencing). It can also expose comprehensive data analysis. Nanopore Direct RNA-Sequencing, on the contrary, has the following benefits: (1) Coupled with fast, streamlined workflows, minimal power quantities allow for highly delicate analysis of gene expression, (2) Transcripts for full-length (the high output of long, full-length reads conveyed by nanopore sequencing grant unambiguous description of splice variations and gene fusions), (3) A precise categorization of the transcript and isoform, (4) Employing direct RNA sequencing to remove PCR bias, (5) Distinguish base alterations alongside nucleotide sequence using direct RNA, (6) Simple recognition of anti-sense transcripts.
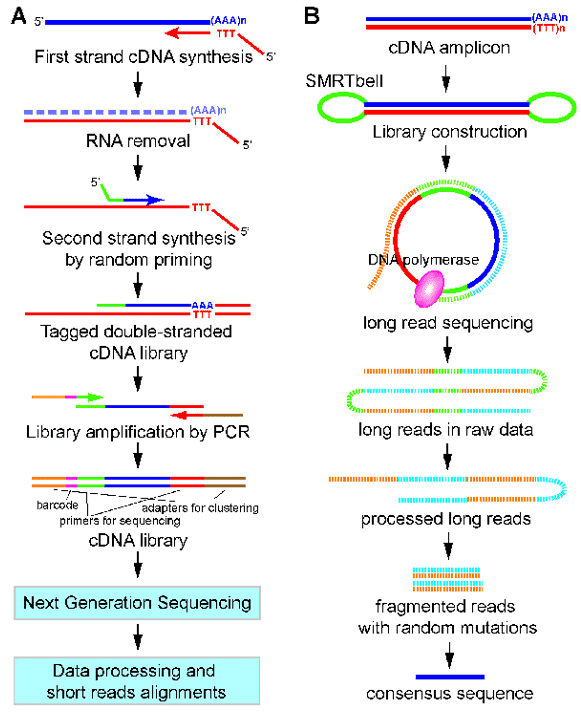
Figure 1. A work flow for 3'end-seq (A) and PacBio Iso-seq (B). (Yeh, 2017)
In terms of function, in the area of medicinal study and agricultural analysis, PacBio Iso-Seq can be utilized. It can be employed in the medical area for transcript annotation, fusion gene exploration, and evaluation of disease mechanisms. It can be utilized for functional research, fusion gene exploration, advancement, and stress test, and coordination for gene prediction and genome annotation in the agricultural field. Nanopore Direct RNA-Seq, on the other hand, can be used to evaluate gene function such as focusing on a specimen having definite functions to expose the primary reason of distinct functions, gene structure like substitute splicing, APA, fusion gene, SSR, CDS prediction, TSS/TES identification, full-length transcript quantification such as locating broad and efficient differential transcripts and recognize functional genes, and RNA methylation such as direct full-length transcriptome sequencing that can identify base alterations at the RNA level, like m6A/m5C.
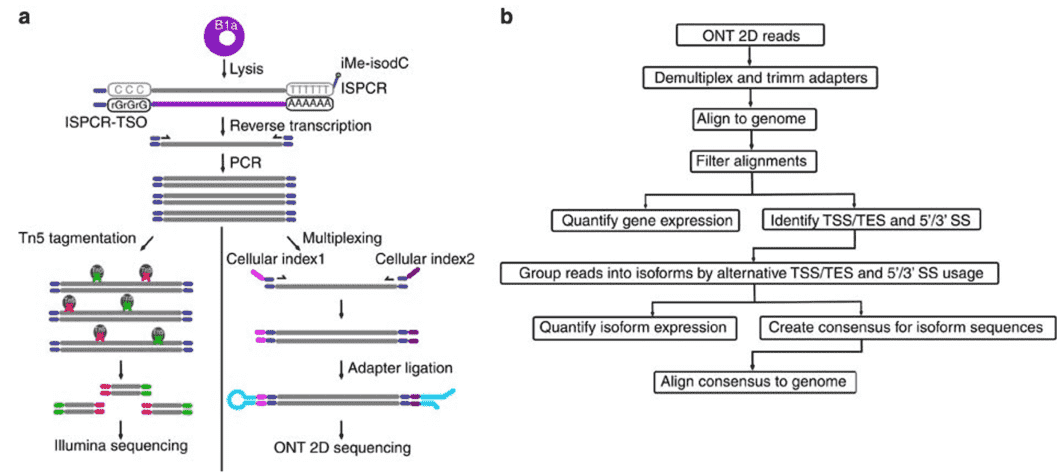
Figure 2. Nanopore Direct RNA-Sequencing. (Byrne, 2017)
References:
- Byrne A, Beaudin AE, Olsen HE, et al. Nanopore long-read RNAseq reveals widespread transcriptional variation among the surface receptors of individual B cells. Nature communications. 2017, 8(1).
- Yeh HS, Zhang W, Yong J. Analyses of alternative polyadenylation: from old school biochemistry to high-throughput technologies. BMB reports. 2017, 50(4).


