
 Sample Submission Guidelines
Sample Submission Guidelines
Epigenomics Sequencing: DNA Methylation Analyses Between Eukaryotes and Prokaryotes
What is DNA Methylation
For more than decades, bases altered by methylation had been thought to exist at a low frequency in DNA. Methyltransferases (DNA methylases) that transmit the chemically active methyl group from S-adenosylmethionine to either carbon 5 of the cytosine solvents or the exocyclic amino group that is affixed to carbon 6 of the adenine residues (m6A) of the DNA chain are involved in this DNA alteration.
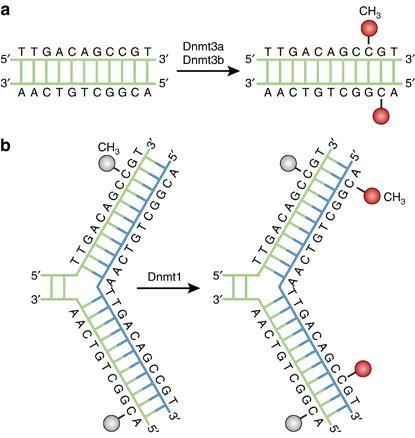 Figure 1. DNA methylation pathways. (Moore et al., 2013)
Figure 1. DNA methylation pathways. (Moore et al., 2013)
The DNA methylation sequence is particular to species. A tiny proportion of the cytosine residues are methylated in the DNA of some prokaryotes, while only adenine residues are methylated in some. The DNA contains both 5-methylcytosine (m5Cyt) and N6-methyladenine (m6Ade) in the third cluster of prokaryotic organisms. Overall, eukaryotic DNA is methylated solely by cytosine residues.
You may interested in
DNA Methylation in Prokaryotes
DNA methylation is utilized in bacteria as a signal for the regulation of a particular DNA-protein interaction. Usually, methylation processes consist of a DNA methylase and one or more proteins binding to DNA that can coincide with the target methylation site on DNA, thereby obstructing that site's methylation.
The target site's methylation restricts protein binding, which can lead to two methylated and nonmethylated alternative methylation states of the target site. This tends to lead to a sequence of DNA methylation that governs which genes are displayed, and hence how the ecosystem engages with a microorganism.
In bacteria, archaea, and phage genomes, there are limited levels of C5-methylcytosine, N4-methylcytosine, and N6-methyladenine. DNA methylation occurs post-replication and is facilitated by DNA methyltransferases that target specific sequences. Prokaryotic DNA methyltransferases fall into two categories: those integrated into restriction-modification systems and standalone enzymes lacking a restriction enzyme counterpart. The primary functions of DNA methylation involve modulating interactions between DNA-binding proteins and their respective target sites. These functions include safeguarding against DNA restriction, facilitating strand discrimination during mismatch repair, regulating the cell cycle, and controlling transcription. DNA methylation also impacts the interactions of bacterial pathogens with their hosts, suggesting potential applications for epigenetic therapies in combating infectious diseases.
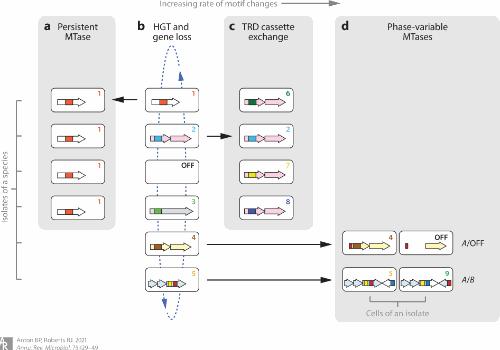 Figure 2. Schematic overview of the evolution of prokaryotic DNA methylation patterns, with four evolutionary mechanisms. (Anton et al., 2021)
Figure 2. Schematic overview of the evolution of prokaryotic DNA methylation patterns, with four evolutionary mechanisms. (Anton et al., 2021)
DNA Methylation in Eukaryotes
DNA methylation in eukaryotes is frequently used to turn off the genes' signaling function. In many eukaryotic mechanisms, such as embryonic growth, genome imprinting, X-chromosome inactivation, and usually maintaining the consistency of chromosomes, DNA methylation has been displayed to have a vital function. In mammals, where the methylation phase for 75% of all CpG dinucleotides is in somatic cells, the process is very precise.
Considering the magnitude of the influence of DNA methylation, it is not strange that several illnesses, such as those noticed in humans are also linked to these effects. The global methylation trend in mammals makes it difficult to assess if methylation is a default situation or is aimed towards particular gene sequences. The CpG islands, however, typically occur near transcription starting sites, implying that there is an identification framework.
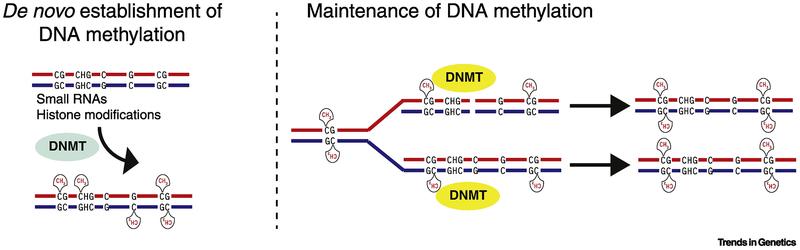 Figure 3. Evolutionary relationship of eukaryotic DNA methyltransferases. (Schmitz et al., 2019)
Figure 3. Evolutionary relationship of eukaryotic DNA methyltransferases. (Schmitz et al., 2019)
Difference Between DNA Methylation in Prokaryotes and Eukaryotes
DNA methylation happens in eukaryotes only on the residues of cytosine and especially for CpG sequences. Whereas in prokaryotes, the main epigenetic signal is the methylation of adenine residues. Only a couple of DNA methyltransferases are used by eukaryotes; in bacteria, where many of them have high sequence precision, the figures are much greater. The significant human gastric pathogen Helicobacter pylori, for instance, has a large DNA methyltransferase gene repertoire, with various strains involving distinct and rather special sequences.
Even so, across prokaryotes and eukaryotes, the defense mechanism of DNA methylation is comparable. In humans and rodents, for instance, embedded viral sequences can be methylated to suppress the genes implemented. In mice as well, the same processes have been discovered to suppress transgenes. The recognition and eradication features of DNA methylation machines, therefore, appear to be preserved.
As the eukaryotic genome is much more intricate in comparison to the prokaryotic genome, the impact of methylated cytosine as a "fifth base" in the eukaryotic genome has been suggested by many research. In addition, many studies showed that the methylation of cytosine is implicated in the eukaryotic genome's functional reorganization.
Table 1 Difference between DNA methylation in prokaryotes and eukaryotes
| Aspect | Prokaryotes | Eukaryotes |
| Type of Methylation | Usually N6-methyladenine (N6-mA) | Usually C5-methylcytosine (C5-mC) |
| Function | Associated with restriction-modification systems, defense against foreign DNA | Involved in gene expression regulation, genome stability, cell differentiation |
| Methyltransferase Type | Often part of restriction-modification systems, specific to double-stranded DNA sequences | More complex, can be specific to sequences but also have broader context methylation |
| Inheritance | Typically vertically inherited, passed to progeny cells through cell division | Inherited through cell division and, in some cases, can be passed to offspring through gametes |
DNA Methylation Sequencing Methods
DNA methylome analysis has been performed using microarrays for many years. Sequencing DNA methylation is a newly developed innovation-based primarily on bisulfite conversion to distinguish between methylated cytosines and unmethylated cytosines. Unmethylated cytosines are transformed to uracils upon bisulfite diagnosis, while 5mCs are non-reactive and maintained. Unmethylated cytosines are read as thymine in the sequencing phase, while methylated cytosines are still read as cytosine.
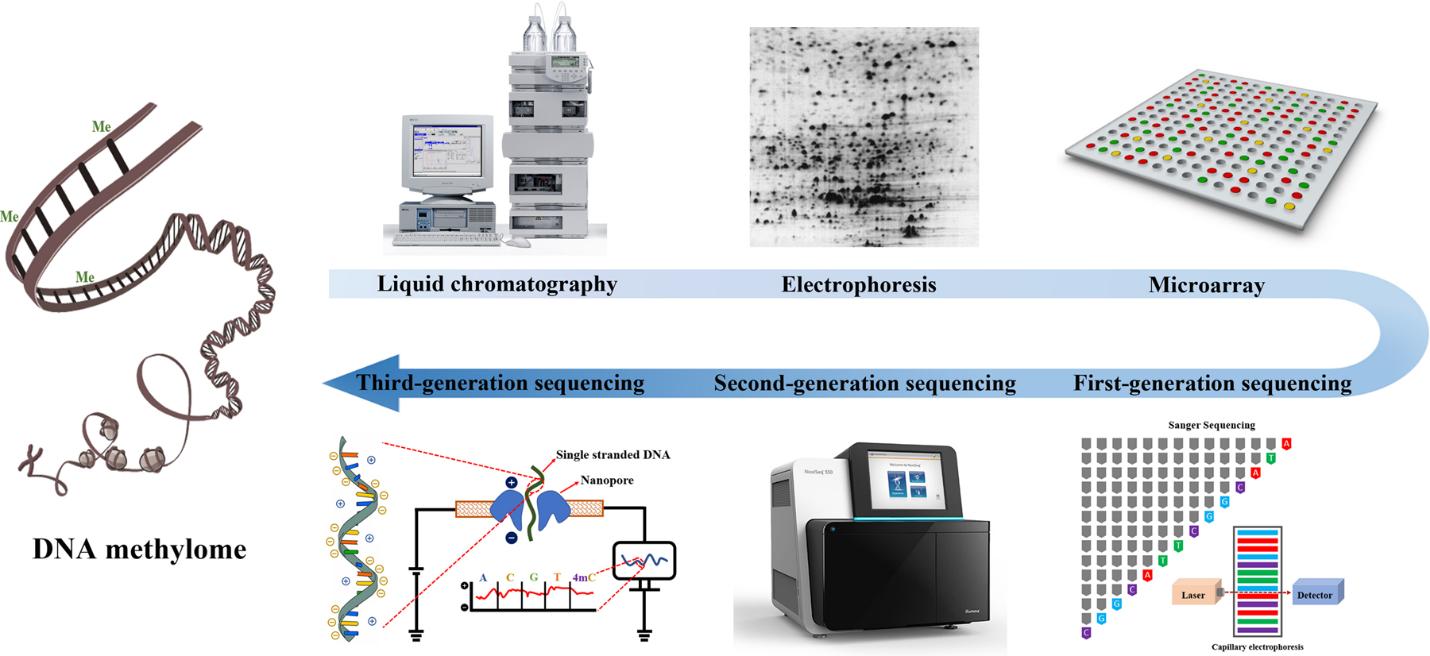 Figure 4. The evolution of DNA methylation profiling methodology. (Li et al., 2021)
Figure 4. The evolution of DNA methylation profiling methodology. (Li et al., 2021)
Service you may intersted in
Bisulfite conversion-based sequencing can be carried out as either Whole-Genome Bisulfite Sequencing (WGBS) or Reduced Representation Bisulfite Sequencing, based on genomic coverage (RRBS). WGBS charges more and there is much greater involvement in the affiliated data evaluation. Instead, RRBS offers a cost-effective method to surveying DNA methylation by sampling genome areas rich in CpG. Genomic DNA is processed with a methylation-insensitive restriction enzyme, such as MspI, to conduct RRBS. To create a library for sequencing, the digested DNA fragments are then confined to adapter ligation, bisulfite conversion, and PCR.
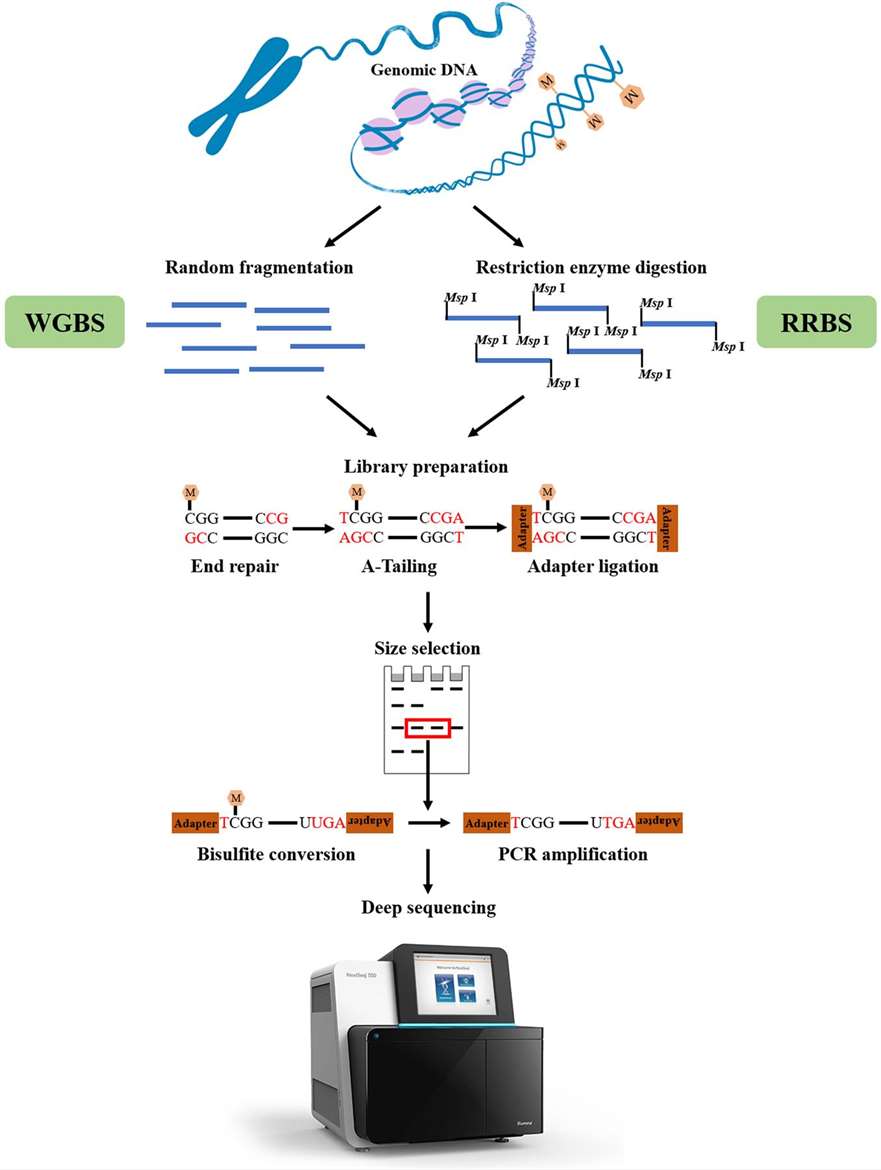 Figure 5. Overview of WGBS and RRBS. (Li et al., 2021)
Figure 5. Overview of WGBS and RRBS. (Li et al., 2021)
WGBS and RRBS are two sequencing techniques relying on bisulfite treatment. Additionally, methodologies that leverage this approach encompass targeted DNA methylation/hydroxymethylation sequencing (oxBS-seq). In oxBS-seq, the conversion of 5-hydroxymethylcytosine (5hmC) to 5-formylcytosine (5fC), and subsequently to uracil (U) post-bisulfite processing, enables the precise identification of 5-methylcytosine (5mC). OxBS-seq presents a simplified experimental workflow compared to BS-seq, allowing for the specific quantification of 5mC and 5hmC at each modified cytosine site in the DNA sample.
The second category comprises sequencing methods based on specific antibody capture, primarily including methylated DNA immunoprecipitation sequencing (meDIP-seq), hydroxymethylated DNA immunoprecipitation sequencing (hmeDIP-seq), and 6mA immunoprecipitation sequencing (6mA-IP-seq). meDIP-seq is a whole-genome methylation detection technique based on the principle of antibody enrichment. It employs methylated DNA immunoprecipitation to selectively enrich DNA fragments methylated on the genome using 5mC antibodies. Subsequently, high-throughput sequencing enables high-precision, CpG-dense, and highly methylated region studies at the whole-genome level. 6mA-IP-seq utilizes antibodies designed for DNA 6mA modification to selectively enrich genomic regions harboring 6mA modification. When coupled with high-throughput sequencing, this approach enables accurate mapping of 6mA modifications on the genome. 6mA-IP-seq presents significant advantages, including high precision, wide detection range, exceptional targeting specificity, and versatile applicability.
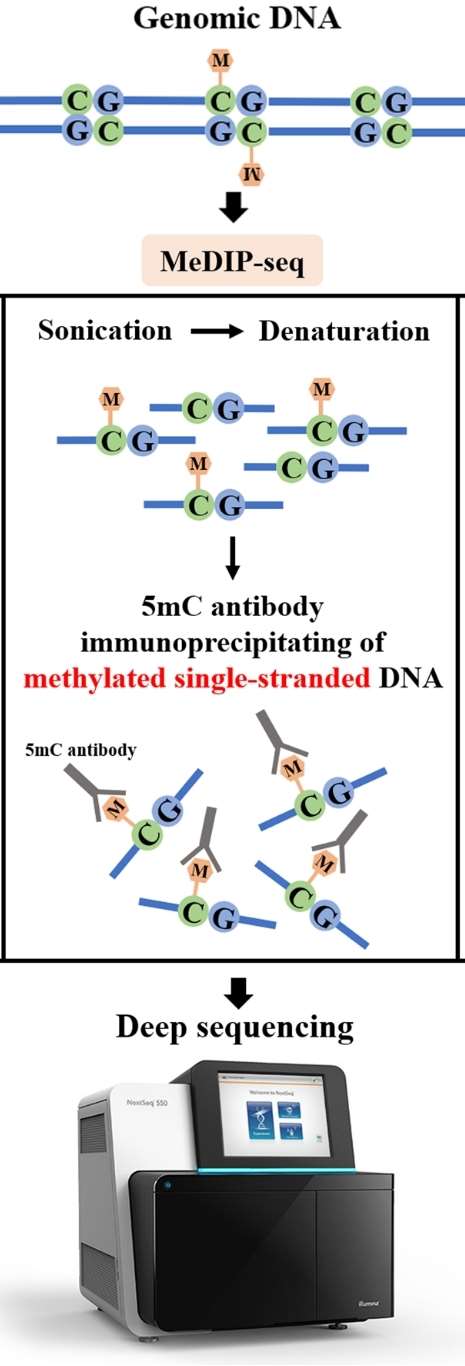 Figure 6. Key principle of MeDIP-seq. (Li et al., 2021)
Figure 6. Key principle of MeDIP-seq. (Li et al., 2021)
Moreover, DNA methylation profiling studies of single cells and minute samples are significantly constrained by library construction sequencing technologies. Traditional library construction methods or single-cell amplification techniques resembling genomic DNA are challenging to apply in methylation experiments. Single-cell whole-genome bisulfite sequencing (scWGBS) addresses this limitation. scWGBS is a highly innovative approach that combines single-cell WGBS library preparation with Illumina next-generation sequencing technology. This method enables visualization of the genome methylation status at single-cell resolution, uncovering cell heterogeneity typically masked by standard bulk methylation sequencing. Methylation studies of single cells and rare samples are primarily applied in tumor mechanisms, cancer research, preimplantation diagnosis, early embryonic development, germ cell recombination, stem cells, and cell heterogeneity research fields.
References:
- Moore, L., Le, T. & Fan, G. DNA Methylation and Its Basic Function. Neuropsychopharmacol. 2013, 38, 23–38.
- Li S, Tollefsbol T O. DNA methylation methods: Global DNA methylation and methylomic analyses. Methods, 2021, 187: 28-43.
- Casadesús J, Sánchez-Romero M A. DNA methylation in prokaryotes[M]//DNA Methyltransferases-Role and Function. Cham: Springer International Publishing, 2022: 21-43.
- Anton B P, Roberts R J. Beyond restriction modification: epigenomic roles of DNA methylation in prokaryotes. Annual Review of Microbiology, 2021, 75: 129-149.
- Schmitz R J, Lewis Z A, Goll M G. DNA methylation: shared and divergent features across eukaryotes. Trends in Genetics, 2019, 35(11): 818-827.


