
 Sample Submission Guidelines
Sample Submission Guidelines
PacBio Sequencing and Droplet-Based MDA Amplification: Pioneering Human Single-Cell Genomics
The field of scientific research has been transformed by the remarkable capabilities of long-read sequencing (LRS). Among the myriad of exciting prospects it offers, one domain stands out in its exploratory phase - human single-cell genomics. This burgeoning research field, with over a decade of history, currently brims with activity and potential. Single-cell genomics, powered by LRS, has the capacity to unravel profound mysteries within cell biology. It can shed light on diverse subjects such as somatic cell genetic variation, tumor evolution, de novo mutation rates, germ cell meiotic recombination, and neurogenetics. Some pioneering studies have already demonstrated the remarkable ability of LRS to identify previously unknown diseases stemming from genetic variations in humans. Moreover, it can uncover clinically significant genetic variants nestled within the enigmatic 'Dark DNA' regions - sections of the human genome that defy analysis through standard short-read sequencing (SRS) methods.
Revolutionizing Single-Cell Genomics with dMDA and PacBio Sequencing
The existing paradigm of single-cell short-read whole genome sequencing leaves a substantial number of variants unexplored, particularly due to their inaccessibility with standard approaches. Additionally, LRS demands a substantial amount of DNA input, posing challenges in its own right. To circumvent issues associated with amplification preferences, chimeric molecules, and allelic deletions, often arising from whole genome amplification (WGA), this study employs an innovative nanoscale droplet multiple-displacement amplification (dMDA) technique.
Methodology
In a nutshell, this study harnessed the potential of fluorescence-activated cell sorting (FACS) to isolate a single cell, releasing its DNA fragments through lysis. These minuscule DNA molecules were then meticulously packaged into approximately 50,000 droplets, each measuring less than 100 µm in diameter. Within these droplets, only one or a few DNA fragments were present, facilitating controlled, limited amplification. Crucially, this approach eliminated the risk of forming intermolecular chimeras. The experiment focused on two distinct CD8+ T-cells, A and B, both sourced from the same human donor. These cells underwent in vitro clonal amplification, succeeded by whole genome amplification (WGA) and sequencing.
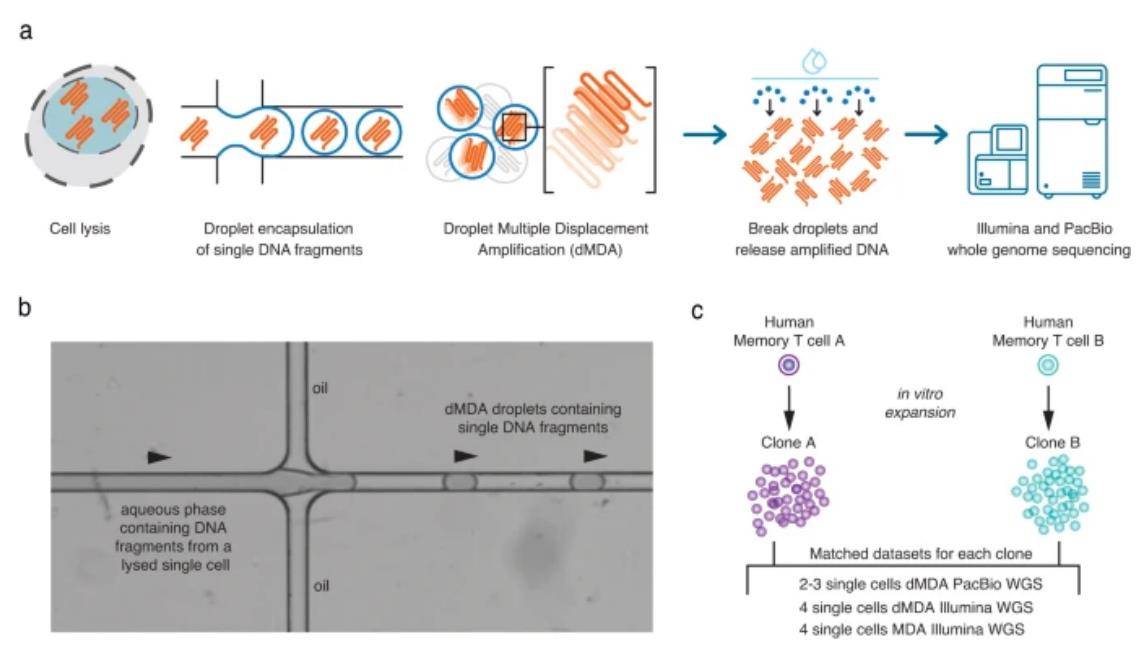 Overview of the single-cell DNA amplification and sequencing experiment. (Hård et al., 2023)
Overview of the single-cell DNA amplification and sequencing experiment. (Hård et al., 2023)
Single-Cell SNV Detection in Long-Read Sequencing Data
In this study, the authors harnessed the power of single-cell sequencing technology, specifically utilizing five dMDA single-cell samples, with two originating from T-cell clone A and three from T-cell clone B. These samples underwent PacBio sequencing, generating an average of 15.7 Gb of data per single cell, while Illumina sequencing produced a substantial 48.7 Gb of data. Both datasets were instrumental for the subsequent identification of single-cell SNVs.
Remarkably, an average of 880,000 SNVs identified in the single-cell PacBio data exhibited concordance with bulk PacBio data, solidifying their authenticity as true SNVs. To draw a meaningful comparison, the authors also subjected Illumina single-cell dMDA and bulk data to the same analysis, resulting in an average of 1.06 million validated SNVs per cell.
Surprisingly, despite PacBio single-cell sequencing yielding only 32% of the data volume generated by Illumina, the number of germline SNVs detected was on par with Illumina's findings. The authors further conducted a comprehensive assessment of SNV calling precision and sensitivity, revealing that, overall, sensitivity was relatively low, especially for the PacBio samples with limited available data. However, PacBio outshone Illumina in terms of SNV identification precision, albeit with slightly lower sensitivity.
Intriguingly, 284,000 high-confidence PacBio SNVs eluded detection in Illumina bulk samples. Among these variants, 6,336 resided in previously designated "dark" gene regions, areas typically inaccessible to standard short-read sequencing methods. Notably, one such region encompassed both introns and exons of NBPF8 and CDC73, the latter existing in the gap left by the Illumina bulk data.
Moreover, beyond germline SNVs, the authors successfully uncovered 27 somatic SNVs within the PacBio data, further enhancing the scope and depth of their investigation.
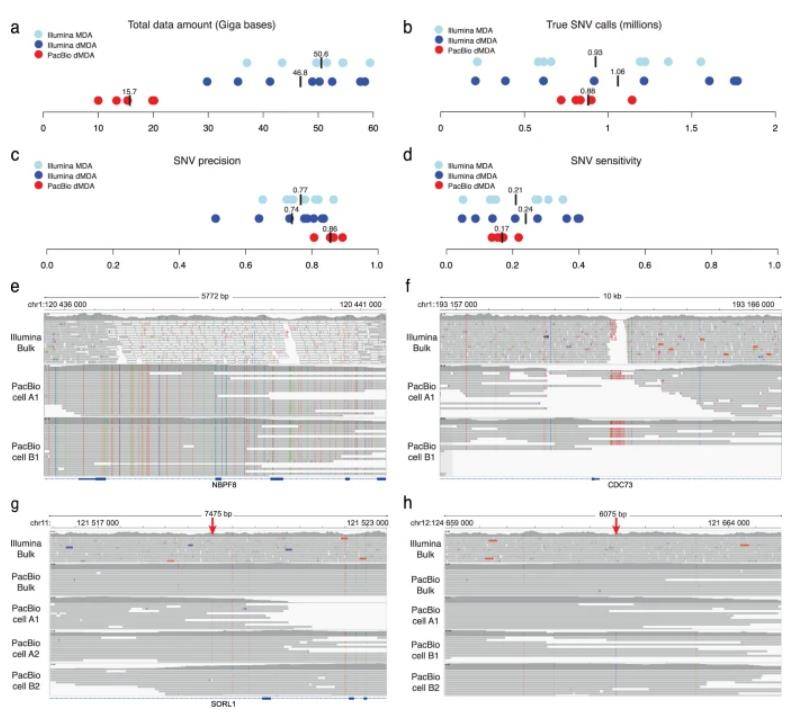 Analysis of SNVs in short- and long-read single-cell data. (Hård et al., 2023)
Analysis of SNVs in short- and long-read single-cell data. (Hård et al., 2023)
Enhancing SV Detection with Long-Read Single-Cell WGS
In this study, researchers utilized Sniffles2 to identify structural variants (SVs) in PacBio data, revealing numerous deletions, insertions, duplications, and inversions per single cell. Over 80,000 unique PacBio SVs, primarily from chimeric dMDA molecules, were absent in bulk samples.
On average, each single cell exhibited 5,473 true SVs, with a majority being deletions and insertions, while duplications and inversions were rare. In contrast, Illumina samples detected only 327 true SVs, significantly fewer.
PacBio's precision for deletions and insertions was 0.73 and 0.66, with slightly higher sensitivity. Duplications and inversions had low precision due to chimeric origins. Notably, PacBio SVs mainly comprised insertions and deletions up to 1 kb, with a peak around 300 bp (ALU repetitive elements) and 6 kb (LINE elements). Some challenging SVs in Illumina data were successfully identified in the single-cell PacBio dataset, including a 710 bp insertion and a 4891 bp deletion.
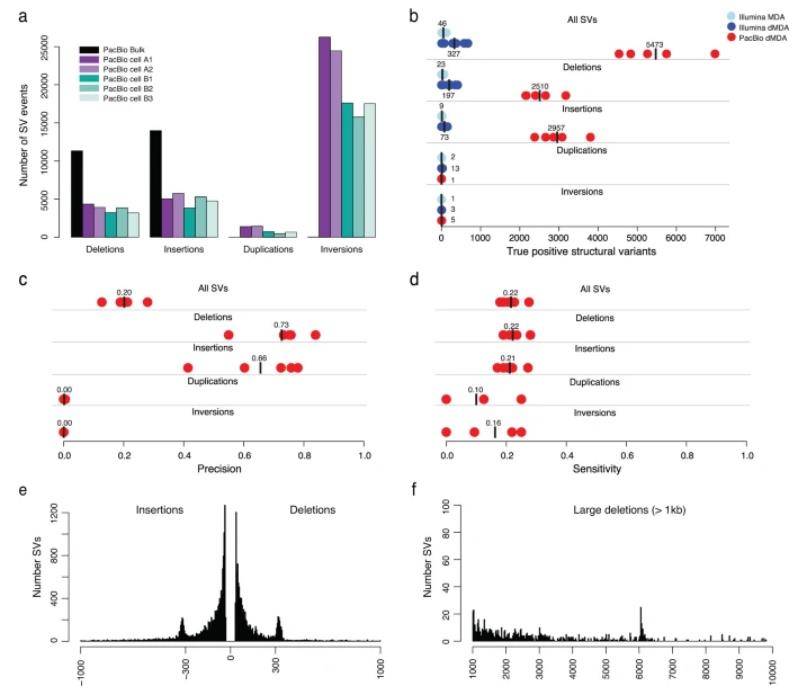 Analysis of SVs in long-read single-cell data. (Hård et al., 2023)
Analysis of SVs in long-read single-cell data. (Hård et al., 2023)
Single-Cell Tandem Repeat Analysis
Using Tandem Genotypes, the authors identified 15,098 TRs initially categorized as pure or heterozygous in PacBio bulk data. On average, 4,770 TR alleles could be accurately genotyped in single cells with profiles similar to the bulk data.
The longest TR observed was 662 base pairs longer than the reference genome, mainly composed of dinucleotide AT sequences—an aspect challenging to resolve in short-read data. While no clear evidence of clonal somatic variation was found in single-cell long-read data, a significant number of repetitive sequences, especially those exceeding 500 base pairs, were absent in single cells due to genotyping failure. This failure often occurred when a sample contained more than two different repeat lengths, making precise TR size determination difficult.
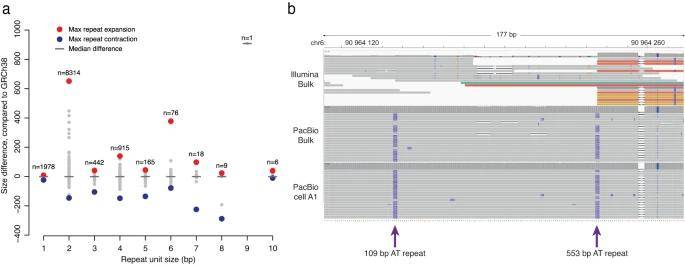 Tandem repeats detected in single-cell long-read data. (Hård et al., 2023)
Tandem repeats detected in single-cell long-read data. (Hård et al., 2023)
Reference:
- Hård, Joanna, et al. "Long-read whole-genome analysis of human single cells." Nature Communications 14.1 (2023): 5164.


