
 Sample Submission Guidelines
Sample Submission Guidelines

What is Whole Genome Sequencing?
Whole Genome Sequencing (WGS) is a cutting-edge, high-throughput technology that reads an organism's entire DNA sequence—including coding genes, non-coding regions, and large-scale structural variations. It captures everything from single-base changes (SNVs) to complex genomic rearrangements (SVs), making it the most thorough method available in modern genomics research.
WGS supports two core approaches:
- De novo assembly: Ideal for newly sequenced species without a reference genome, helping to resolve complex repeat regions.
- Resequencing: Detects SNPs, insertions/deletions (InDels), and structural changes based on a known reference genome.
Unlike probe-based methods, WGS provides unbiased, uniform genome-wide coverage—critical for analyzing non-coding regions, repetitive sequences, and structural variations that often go undetected in targeted approaches.
CD Genomics delivers precision WGS solutions across multiple platforms, including Illumina, PacBio, and Oxford Nanopore, to meet the diverse needs of species-specific studies and research goals.
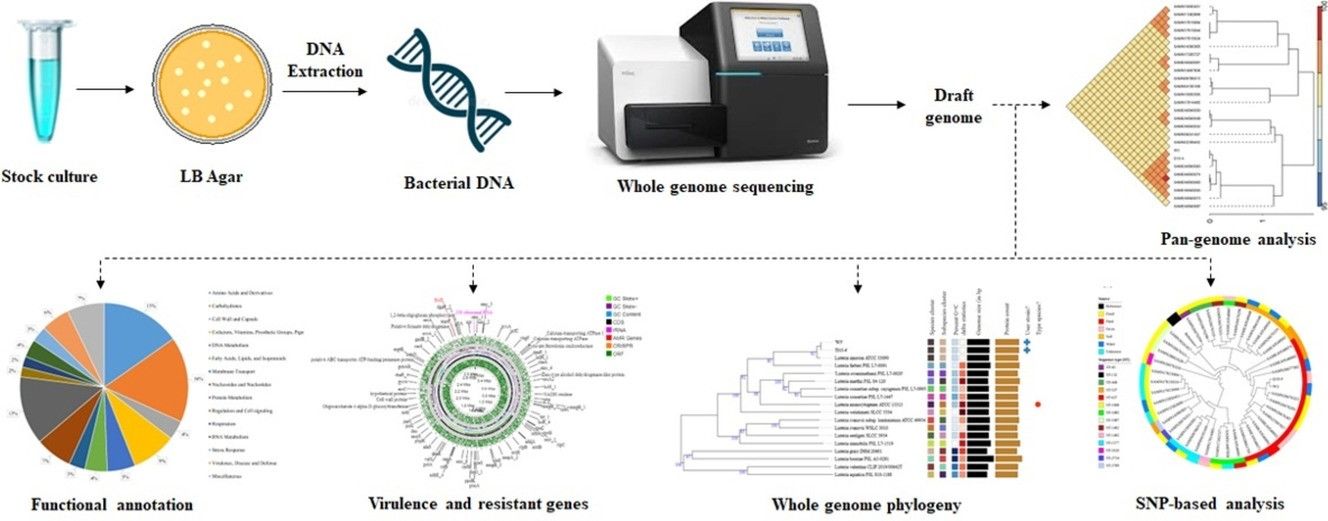 Whole genome sequencing uncovers virulence factors, mobile genetic elements, and potential environmental transmission of bacterial strains in cattle farm settings. (Rivu, Supantha, et al., 2024)
Whole genome sequencing uncovers virulence factors, mobile genetic elements, and potential environmental transmission of bacterial strains in cattle farm settings. (Rivu, Supantha, et al., 2024)
Why Choose Whole Genome Sequencing?
WGS is rapidly becoming an indispensable tool in life sciences research due to its unbiased nature, comprehensive scope, and high-resolution capabilities. Unlike traditional targeted sequencing, chips, and exome sequencing, WGS overcomes technological limitations, offering deeper and more nuanced data support for scientific exploration.

- Unbiased Coverage: WGS transcends the constraints of targeted technologies, capturing SNPs, InDels, structural variations (SVs), and non-coding region mutations in one go, ensuring that critical genetic markers are not overlooked.
- Versatility Across Research Areas: It provides precise solutions for research across diverse species, including plants, animals, microbes, and even ancient DNA, effectively tackling challenges like repetitive sequences and highly polymorphic regions.
- Cost-Effective Strategies: With flexible coverage depths ranging from 0.1x to 100x, WGS allows cost savings through low-pass screening and facilitates in-depth sequencing for rare variant discovery, with data that can be reused indefinitely.
- Multi-dimensional Data Integration: By combining with epigenetics (5mC native detection), WGS unveils functional mechanisms of variations, accelerating the conversion of sequence data into biological insights.
| Limitations of Traditional Techniques | Core Advantages of WGS |
|---|---|
| Can only detect known SNP sites | Discovers novel rare variants and sample-specific mutations |
| Ignores non-coding regions and regulatory sequences | Provides whole-genome coverage, including both coding and non-coding areas |
| Capture efficiency relies on probe design, uneven coverage | Utilizes PCR-Free library preparation, achieving uniformity fluctuation within ±5% |
| Difficulty in identifying SVs and large segment rearrangements | SV detection accuracy exceeds 95%, ideal for researching complex variations |
Whole Genome Sequencing Service Options
CD Genomics offers a variety of flexible whole genome sequencing service types to cater to different research objectives and budget requirements:

Standard Whole Genome Sequencing
High coverage | Comprehensive variant profiling | Flexible design
Detailed Parameters ↓
Whole Genome Re-Sequencing Service
Reference-based analysis | Detect SNVs, InDels, CNVs
Explore Re-Sequencing Service →
Plant/Animal Whole Genome de novo Sequencing
No reference genome required | Chromosome-scale assembly | Multi-platform strategy
Explore de novo Genome Sequencing→
De Novo Whole Genome Sequencing Service
No reference needed | Complete genome assembly
View De Novo WGS Service →
Human Whole Genome PacBio SMRT Sequencing
Long-read sequencing | Resolve complex regions
See Human PacBio WGS Details →
Bacterial Whole Genome de novo Sequencing
Complete genome assembly | Microbial insights
Explore Bacterial Genome Sequencing →
Fungal Whole Genome de novo Sequencing
High-complexity genome assembly | Functional genomics
Learn More About Fungal WGS →
Microbial Whole Genome Sequencing
Broad microbial targets | Accurate identification
Discover Microbial WGS Solutions →
Shallow Whole Genome Sequencing
Low-pass WGS | CNV analysis | Population stratification
Learn More About Shallow WGS →Whole Genome Sequencing Service Workflow
At CD Genomics, we offer a seamless, end-to-end whole genome sequencing service designed to ensure consistent, high-quality results. Our standardized workflow—from sample submission to data delivery—is built to support reproducibility, streamline research, and accelerate discovery across all types of genomic studies.

Whole Genome Sequencing Strategies
Sequencing Platforms and Read Lengths:
- Illumina NovaSeq 6000 / X: Delivers 150 bp paired-end reads with high throughput, ideally suited for resequencing and large-scale sample analyses.
- PacBio Sequel IIe: Provides average high-fidelity long reads ranging from 15–25 kb, perfect for de novo assembly and complex structural region analysis.
- Oxford Nanopore PromethION: Capable of read lengths exceeding 100 kb, suitable for assembling ultra-long fragments and detecting structural variations.
Optional Strategies:
- Coverage Depth: Standard depth (30×), High depth (over 60×), Low depth (1–5×)
- Analysis Methods: Resequencing, De novo assembly, Hybrid strategies (combining short and long reads)
- Customization: Tailored project design and analysis processes
Library Construction Methods:
- Standard or PCR-Free Libraries: Improve coverage uniformity and reduce GC bias
- Long Fragment Libraries (PacBio/ONT): Enhance assembly quality in complex or repetitive genomes
Supported Sample Types:
- High-quality genomic DNA
- Blood, cultured cells, fresh/frozen tissues, FFPE specimens
For tailored whole genome sequencing solutions or any inquiries regarding sequencing strategies, please contact our expert team to receive professional guidance and support.
Whole Genome Sequencing Bioinformatics Analysis
CD Genomics offers comprehensive and flexible bioinformatics analysis services, ranging from basic data processing to advanced customized analyses. Our solutions facilitate in-depth exploration of genomic variations and functionalities.
Basic Analysis Modules:
- Raw Data Quality Control and Filtering: Ensures data integrity and reliability for downstream analyses.
- Alignment to Reference Genome (or De Novo Assembly): Establishes a foundation for identifying genetic variations.
- Variant Detection: Identifies SNVs, InDels, CNVs, and SVs.
- Genome Coverage and Depth Statistics: Provides insights into the sequencing completeness and depth of analysis.
- Functional Annotation and Mutation Classification: Categorizes and annotates detected variants for further interpretation.
Advanced Analysis Modules (Customizable):
- Candidate Pathogenic Gene and Pathway Enrichment Analysis: Highlights potential genes and pathways implicated in disease.
- Familial Genetic Pattern and Linkage Analysis: Investigates hereditary patterns and genetic linkages within families.
- Population Structure, SNP Frequency Distribution, and Fst Calculation: Explores genetic diversity and population differentiation.
- Fusion Gene Detection and Viral Integration Analysis (for microbial or specific needs): Identifies genetic fusions and viral DNA integrations.
- Structural Variation Visualization and Re-arrangement Mapping: Visualizes complex genetic structures and rearrangements.
For personalized bioinformatics analysis or specific research needs, please reach out to our experts for professional advice and support tailored to your project's requirements.
Applications of Whole Genome Sequencing
WGS is a trusted method for obtaining complete and in-depth genetic information, and it finds application across various research fields. It empowers researchers to comprehensively analyze genome structures and variations, fitting a range of scientific inquiries, including but not limited to:
- Population Genetics and Evolutionary Analysis
- Precisely uncovers population genetic structures, phylogenetic relationships, and selection signals, facilitating studies on population evolution and species differentiation.
- Complex Traits and Disease Association Studies
- Utilized in Genome-Wide Association Studies (GWAS) and Quantitative Trait Loci (QTL) mapping, aiding in the discovery of key genetic variations linked to phenotypes.
- Plant and Animal Genome Construction and Breeding Improvement
- Supports the development of high-quality reference genomes, identification of significant functional genes, and accelerates the molecular breeding process.
- Microbial Genome Research and Surveillance
- Applied in de novo pathogen sequencing, research on resistance mechanisms, strain tracing, and public health monitoring.
- Non-Model Organism Research
- Rapidly acquires whole genome information of new species, supporting ecological adaptability, species conservation, and genetic resource development.
- Functional Gene and Regulatory Region Exploration
- Encompasses non-coding regions to capture regulatory elements and epigenetic regulatory variations, supporting functional gene research.
- Exogenous Insertion and Viral Integration Detection
- Detects viral integrations or transgene insertion events in genomes, suitable for the tracking of exogenous sequences and safety assessments.

Sample Requirements for Whole Genome Sequencing
| Sequencing Type | Total Genomic DNA Requirement | Minimum Usable Amount | DNA Concentration Requirement | Purity Requirement (OD260/280) | Notes |
|---|---|---|---|---|---|
| Whole Genome Sequencing | ≥ 500 ng | 200 ng | ≥ 10 ng/μL | 1.8 ~ 2.0 | Suitable for routine whole genome sequencing |
| Whole Genome Sequencing (PCR-Free) | ≥ 1 μg | 500 ng | ≥ 20 ng/μL | 1.8 ~ 2.0 | Avoids PCR amplification bias, ensures higher data uniformity |
| Whole Genome Sequencing (PacBio) | ≥ 1 μg | — | ≥ 80 ng/μL | 1.8 ~ 2.0 | Ideal for long-read sequencing, requires high DNA concentration |
| Whole Genome Sequencing (Nanopore) | ≥ 5 μg | — | ≥ 20 ng/μL | 1.8 ~ 2.0 | Suitable for ultra-long-read sequencing, requires a large amount of DNA |
- All DNA samples must undergo purity and concentration testing to ensure sequencing quality.
- If you have questions regarding sample preparation or require a custom plan, feel free to contact us anytime for expert assistance.
Why Choose CD Genomics for Whole Genome Sequencing?
From advanced sequencing platforms to high-quality data delivery, CD Genomics offers an efficient, end-to-end WGS solution tailored to diverse research needs. Whether you're studying rare variants or sequencing ancient DNA, our team ensures reliable results with flexible support.
- Multi-platform Integration: Harness the combined strengths of Illumina, PacBio, and Nanopore technologies to suit any project—from variant detection to de novo assembly.
- Ultra-High Throughput: Our HiSeq X Ten system processes up to 600 samples per day, delivering 30× coverage data efficiently.
- Exceptional Accuracy: PacBio HiFi reads achieve Q33 (>99.95% base call accuracy), increasing structural variant detection sensitivity by 300%.
- Cross-Species Support: Proven success rates >98% for human, plant, animal, microbial, and ancient DNA samples.
- Custom Solutions: Flexible analysis workflows for special cases like viral insertion detection or low-quality samples.
- End-to-End Support: Get expert guidance at every stage—project design, workflow monitoring, and post-analysis consultation.

Reference
- Rivu, Supantha, Abiral Hasib Shourav, and Sangita Ahmed. "Whole genome sequencing reveals circulation of potentially virulent Listeria innocua strains with novel genomic features in cattle farm environments in Dhaka, Bangladesh." Infection, Genetics and Evolution 126 (2024): 105692. https://doi.org/10.1016/j.meegid.2024.105692
- Nakagawa, Hidewaki, and Masashi Fujita. "Whole genome sequencing analysis for cancer genomics and precision medicine." Cancer science 109.3 (2018): 513-522.
- https://doi.org/10.1111/cas.13505
- Tyler, A.D., Mataseje, L., Urfano, C.J. et al. Evaluation of Oxford Nanopore's MinION Sequencing Device for Microbial Whole Genome Sequencing Applications. Sci Rep 8, 10931 (2018). https://doi.org/10.1038/s41598-018-29334-5
- Turro, E., Astle, W.J., Megy, K. et al. Whole-genome sequencing of patients with rare diseases in a national health system. Nature 583, 96–102 (2020). https://doi.org/10.1038/s41586-020-2434-2
- Kosugi, S., Momozawa, Y., Liu, X. et al. Comprehensive evaluation of structural variation detection algorithms for whole genome sequencing. Genome Biol 20, 117 (2019). https://doi.org/10.1186/s13059-019-1720-5
Demo Results
Partial results are shown below:

Distribution of base quality.

Distribution of base content.

Shared SNP number between samples.

SNP mutation type distribution.
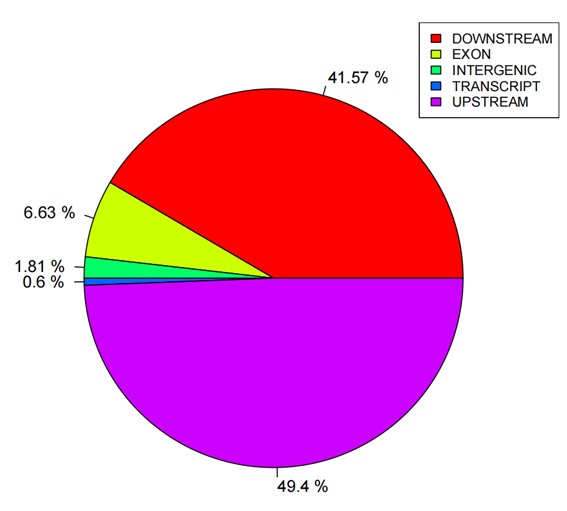
Statistics pie of SNP annotations.

Shared InDel number between samples.

InDel length distribution in both the whole genome scale and CDS regions.

Statistics pie of InDel annotations.
Whole Genome Seq FAQs
1. What's the recommended sequencing depth for human WGS?
We recommend 30× depth (~90 Gb of raw data) for general variant detection and resequencing. For detecting low-frequency mutations, higher depth (e.g., 60×) is more suitable.
2. Can FFPE samples be used for WGS?
Yes, but with caution. FFPE DNA often shows degradation and artefacts. To improve outcomes:
- Use fresh 10 µm sections containing tissue.
- Ensure at least 1 cm² total tissue area.
- Consider using optimized library prep kits specifically for FFPE.
3. What types of variants can WGS detect?
WGS captures a broad spectrum of genomic variations in one pass:
- Single nucleotide variants (SNVs)
- Small insertions/deletions (InDels)
- Copy number variants (CNVs)
- Structural variants (SVs)
We also provide functional annotations to help assess biological impact.
4. My sample is low-quantity or degraded—can it still be sequenced?
It may still be viable. We offer low-input and damage-tolerant library prep solutions. Contact us for a personalised feasibility assessment.
5. How do I choose the right sequencing strategy?
It depends on your research goal:
- Resequencing → Illumina
- De novo assembly or SV analysis → PacBio/Nanopore
Need help? Our experts can recommend the optimal platform and prep strategy.
6. How should I choose sequencing depth?
Standard: 30× for variant detection.
Advanced: ≥60× or hybrid (short + long reads) for complex rearrangements or assembly tasks.
We tailor depth and strategy to your specific project.
7. How can we ensure the reliability of genome assembly results?
To evaluate the integrity and reliability of a genome assembly, multiple metrics and validation methods are employed:
- Contig N50 and Scaffold N50: These metrics indicate the continuity of assembled sequences and are commonly used to assess assembly completeness.
- Transcriptome Alignment: EST datasets or RNA-seq reads can be aligned to the assembly to evaluate gene coverage and continuity.
- Conserved Genes: Benchmarking Universal Single-Copy Orthologs (BUSCO) analysis is often used to assess the completeness of conserved genes.
- BAC Clone Comparison: Bacterial artificial chromosome (BAC) sequences can serve as high-confidence references to validate assembly accuracy at the structural level.
8. How do you handle highly repetitive and heterozygous regions in genome assembly?
Repetitive sequences are prevalent across a wide range of species—from microbes to mammals—and present a significant challenge for accurate assembly. Similarly, heterozygosity complicates haplotype resolution in diploid and polyploid organisms. To address these complexities:
- We employ a hybrid sequencing strategy, integrating high-accuracy short reads from Illumina HiSeq, long reads from PacBio, and in some cases, legacy Sanger reads.
- This approach improves both the resolution of repeat regions and the phasing of heterozygous alleles, ensuring greater structural and allelic accuracy in the final assembly.
9. How is genome size estimated?
Several approaches are available to estimate the genome size prior to sequencing:
- Online Databases: For well-studied species, databases such as Animal Genome Size Database provide curated genome size estimates.
- Flow Cytometry: A standard method that estimates genome size by measuring DNA content in stained cells.
- Genome Survey via K-mer Analysis: This computational method uses short-read data to estimate genome size, repeat content, and heterozygosity by analyzing the frequency distribution of k-mers (subsequences of length k) in the sequencing data.
10. Can I order sequencing without bioinformatics analysis?
Absolutely. Choose sequencing only, analysis only, or a full-service package—whichever suits your workflow best.
11. Is project progress tracked and reported?
Yes. Each project is assigned a dedicated manager and support team. You'll receive timely updates at every key milestone.
Whole Genome Seq Case Studies
Customer Publication Highlight
Genetic mapping of the Rcs2 locus in soybean cultivar Kent for resistance to frogeye leaf spot
Journal: Crop Science
Impact Factor: ~2.8 (2022)
Published: 2023
DOI: 10.1002/csc2.21043
Background
Frogeye leaf spot (FLS), caused by Cercospora sojina, leads to yield losses of up to 30% in susceptible soybean cultivars. The Rcs2 locus in soybean cultivar Kent confers resistance to all known U.S. races of C. sojina. However, the genomic basis of Rcs2 remained unmapped, hindering its application in marker-assisted breeding. This study aimed to molecularly map Rcs2, identify candidate genes, and develop robust molecular markers.
Project Objectives
- Genetic Mapping: Precisely locate the Rcs2 locus on the soybean genome.
- Candidate Gene Validation: Narrow the locus to functional genes linked to resistance.
- Marker Development: Design KASP markers for accelerated breeding programs.
CD Genomics' Services
As a leading genomics partner, CD Genomics delivered:
1. Whole Genome Sequencing (WGS)
- Platform: Illumina NovaSeq X (150 bp paired-end reads).
- Coverage: 30x depth for parental lines (Kent, Forrest) and recombinant inbred lines (RILs).
- Library Preparation:
- DNA shearing via Covaris g-TUBE (~470 bp fragments).
- AMPure XP Beads for size selection.
- Dual-indexed libraries for multiplexed sequencing.
- Quality Control: FastQC v0.11.9 for raw read evaluation.
- Alignment: Bowtie2 v2.4.1 against the Williams82.a2.v1 reference genome.
- Variant Detection: BCFtools v1.10.2 for SNP/InDel identification.
3. Marker Development
- KASP Assays: Designed SNP markers (e.g., GSM783, GSM990) within the Rcs2 locus.
Key Findings
1. Precision Mapping of Rcs2
- Locus Localization:
- BSA and linkage analysis narrowed Rcs2 to 336 kb on chromosome 11 (32.2–32.5 Mb).
- Identified 11 candidate genes with polymorphisms in Kent, including LRR receptor-like kinases and amino acid transporters.
2. High-Accuracy Markers
- Validation: KASP marker GSM783 achieved 94% accuracy in differentiating resistant/susceptible RILs.
- Phenotypic Correlation: RILs with the resistant allele exhibited 33% lower disease severity (P < 0.001).
3. Resistance Mechanism
- Candidate Genes:
- Glyma.11g228300 (amino acid transporter) and Glyma.11g230200 (transcription factor) showed non-synonymous mutations in Kent.
- Promoter variations in LRR-RLKs suggest roles in pathogen recognition signaling.
Figures Referenced
 Genomic regions identified using a bulked segregant analysis on (a) chromosome 11 and (b) chromosome 16 for resistance to frogeye leaf spot in the F2:3 population.
Genomic regions identified using a bulked segregant analysis on (a) chromosome 11 and (b) chromosome 16 for resistance to frogeye leaf spot in the F2:3 population.
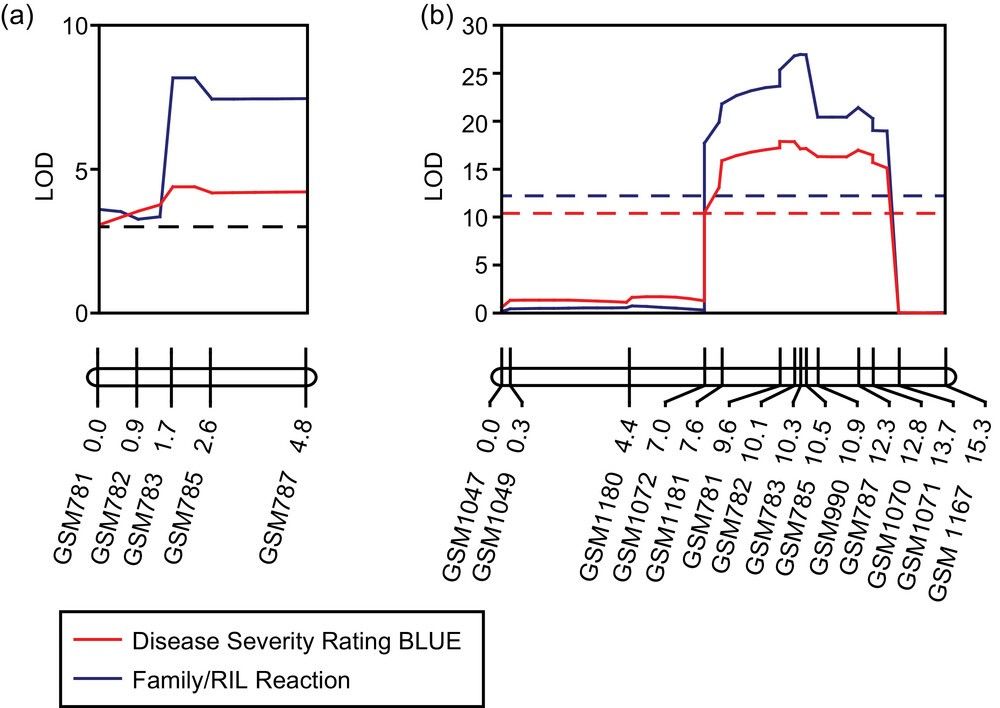 Linkage maps and graphs for the Rcs2 locus on chromosome 11 in the (a) F2:3 and (b) recombinant inbred line (RIL) populations.
Linkage maps and graphs for the Rcs2 locus on chromosome 11 in the (a) F2:3 and (b) recombinant inbred line (RIL) populations.
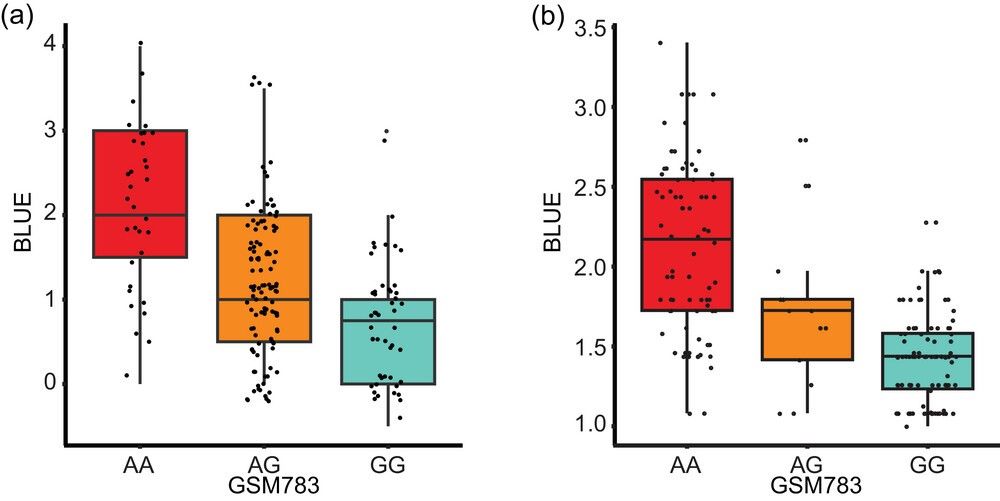 Association of single nucleotide polymorphism (SNP) marker GSM783 with visual disease severity rating best linear unbiased estimates (BLUE) in (a) F2:3 and (b) recombinant inbred line populations.
Association of single nucleotide polymorphism (SNP) marker GSM783 with visual disease severity rating best linear unbiased estimates (BLUE) in (a) F2:3 and (b) recombinant inbred line populations.
Implications
This study resolves the genetic basis of Rcs2-mediated resistance, enabling:
- Marker-Assisted Selection (MAS): KASP markers (GSM783/GSM990) streamline breeding for FLS resistance.
- Durable Resistance: Pyramiding Rcs2 with other loci (e.g., Rcs3) enhances resilience against evolving C. sojina races.
- Functional Genomics: Candidate genes provide targets for CRISPR validation and mechanistic studies.
Related Publications
Here are some publications that have been successfully published using our services or other related services:
Identification of factors required for m6A mRNA methylation in Arabidopsis reveals a role for the conserved E3 ubiquitin ligase HAKAI
Journal: New phytologist
Year: 2017
High-Density Mapping and Candidate Gene Analysis of Pl18 and Pl20 in Sunflower by Whole-Genome Resequencing
Journal: International Journal of Molecular Sciences
Year: 2020
Isolation and Characterization of Bacteria Associated with Onion and First Report of Onion Diseases Caused by Five Bacterial Pathogens in Texas, U.S.A.
Journal: Plant Disease
Year: 2023
Generation of a highly attenuated strain of Pseudomonas aeruginosa for commercial production of alginate
Journal: Microbial Biotechnology
Year: 2019
Combinations of Bacteriophage Are Efficacious against Multidrug-Resistant Pseudomonas aeruginosa and Enhance Sensitivity to Carbapenem Antibiotics
Journal: Viruses
Year: 2024
Identification of the genetic elements involved in biofilm formation by Salmonella enterica serovar Tennessee using mini-Tn10 mutagenesis and DNA sequencing
Journal: Food Microbiology
Year: 2022
See more articles published by our clients.


