
 Sample Submission Guidelines
Sample Submission Guidelines
A Guide to De Novo Genome Assembly
Genome assembly is one of the main purposes of sequencing. De novo genome assembly is a strategy for genome assembly, representing the genome assembly of a novel genome from scratch without the aid of reference genomic data. De novo genome assemblies assume no prior knowledge of the source DNA sequence length, layout or composition.
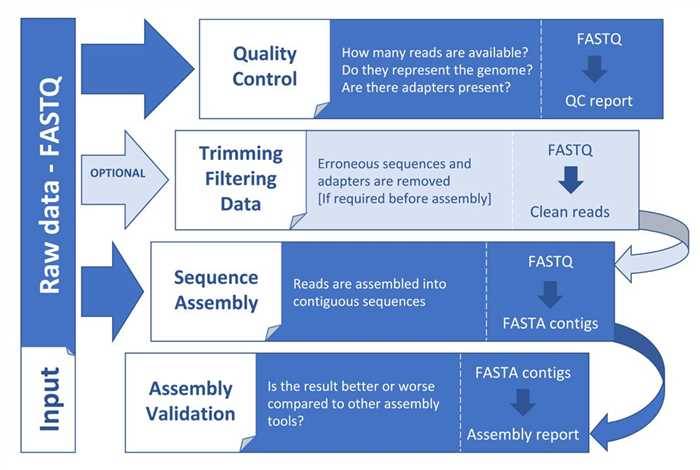 Figure 1. General steps in a genome assembly workflow (Angel et al. 2018).
Figure 1. General steps in a genome assembly workflow (Angel et al. 2018).
Read types
To assemble a genome with the next generation sequencing platforms, computer programs typically use single and paired reads. These ‘reads’ vary from 20 to 1000 bp in length depending on the sequencing platform used. Single reads are the short sequenced fragments, which can be joined up according to overlapping regions into a continuous sequence known as a ‘contig’. Paired reads are about the same length as single reads, but coming from either end of DNA fragments. Compared to single reads, paired reads are preferred, because they help link contigs into ‘scaffolds’ and indicate the size of repetitive regions.
Even so, repetitive sequences, variants, missing data and mistakes sometimes limit the efficiency and accuracy of genome assembly. Long reads technologies emerge at the right moment, which span stretches of repetitive regions and thus generate a contiguous reconstruction of the genome. Currently, this new generation is dominated by two methods, single molecule real-time (SMRT) sequencing and nanopore sequencing, championed by Pacific Biosciences (PacBio) and Oxford Nanopore Technologies, respectively. SMARTdenovo is a long read OLCassembly pipeline that has been shown to produce assemblies of reasonably high continuity from both MinION and SMRT reads.
Illumina Genome assembly
We will take the Illumina genome assembly as an example to introduce the workflow of genome assembly with NGS data, since Illumina sequencing is one of the most common approaches for genomics studies.
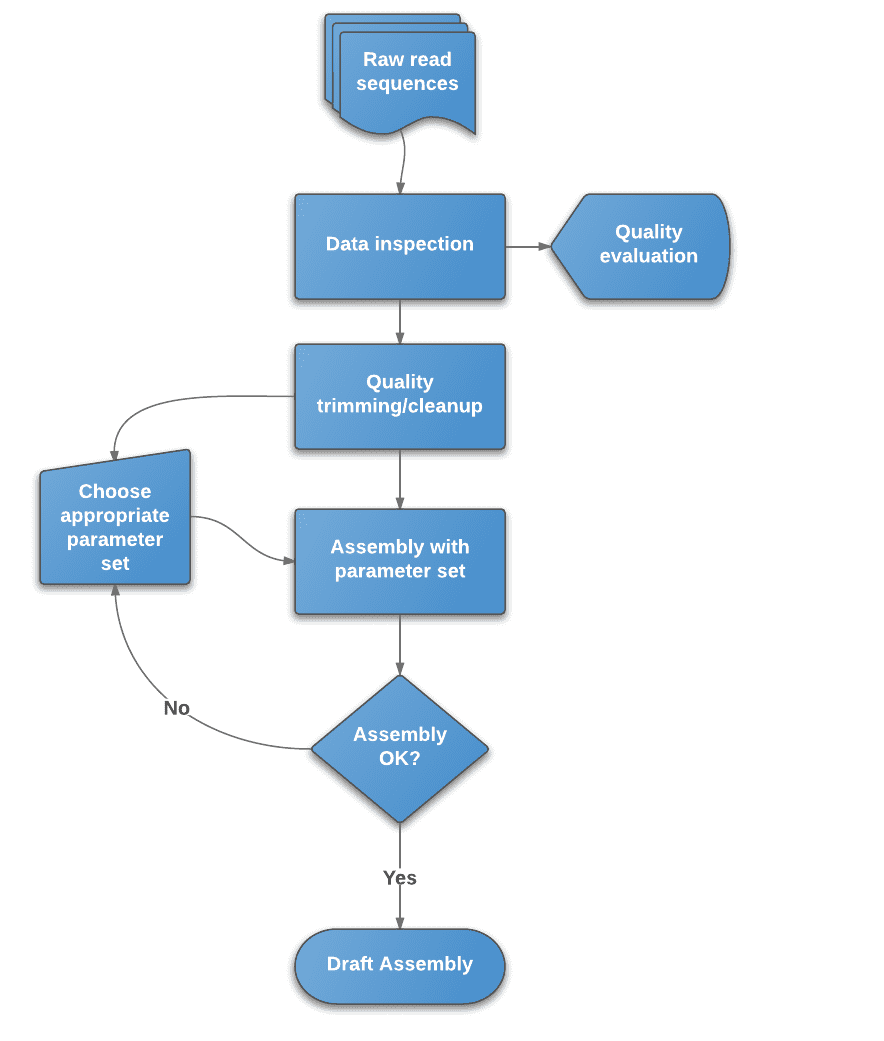 Figure 2. Flowchart of de novo assembly protocol.
Figure 2. Flowchart of de novo assembly protocol.
- Assessing the quality of reads
Prior to genome assembly, it is important to assess the quality of the sequence data, as it may lead to erroneous conclusions. The reads can be stored in Fasta, FastQ, SAM, and BAM formats. The FastQ is the most common read file, as it is produced by the Illumina sequencing pipeline. In addition to read types, other issues including the number of reads, the GC content, and contamination, are also need to be considered.
Base calling accuracy evaluates the probability of that a given base is called incorrectly, and is commonly determined by the Phred quality scores (Q score). FastQC is the most common tool for quality control of raw data. Main outputs of FastQC include read length, quality encoding type, %GC, total number of reads, the presence of highly recurring k-mers, the presence of large numbers of N’s in reads, and dips in quality near the beginning, middle, or end of the reads.
- Pre-processing of raw data
Once the sequence data quality is determined, many tools for quality trimming are available in Galaxy or by command line, such as Trimmomatic. It can handle read pairing, if you’ve got paired reads. Trimmomatic can perform multiple read trimming functions sequentially, including:
- Adapter trimming. This function trims adapters, barcodes, and other contaminants.
- Sliding window trimming. This function works to measure the average quality and trims accordingly.
- Bases quality trimming. This function trims trailing and leading bases of poor quality.
- Minimum read length. This function makes sure that the reads after all trimming steps are longer than the minimum read length. If not, the reads are removed.
PRINSEQ is a similar tool for quality trimming of raw data.
- De novo genome assembly
The next step is to assemble the quality trimmed reads into draft contigs. The suggested assembly software for this step is the Velvet Optimiser which wraps the Velvet Assembler. The Velvet Assembler specifically written for Illumina style short reads, and it uses the de Bruijun graph approach. Velvet Assembler and Velvet Optimiser can take multiple read files (such as SAM, BAM, FastQ, and Fasta) and types (such as single ended, paired end, and mate pair). The quality of contigs assembled by Velvet is primarily dependent on its parameter setting. The most critical parameters include the hash size, the expected coverage, and the coverage cutoff. Alternative de novo assemblers include Spades, SOAP-denovo, MIRA, and ALLPATHS.
- Assembly polishing
After all the steps above, you will get draft contigs containing some gaps or regions of ‘N’s. And some of them may be mis-assemblies. Next, you need to improve your assembly using mis-assembly checking and assembly metric tools, such as QUAST, InGAP-SV, and Mauve assembly metrics.
If you want to finish your genome, you need to use more different data, or use other tools with your current data. And the alternative genome finishing tools include semi-automated gap fillers (e.g. Gap filler), genome visualisers and editors (Artemis, IGV, Geneious, CLC BioWorkbench), and annotation tools (e.g. Prokka, RAST, and JCVI Annotation Service).
References:
- Lannoy C D, Ridder D D, Risse J. The long reads ahead: de novo genome assembly using the MinION. F1000Research, 2017, 6.
- Baker M. De novo genome assembly: what every biologist should know. Nature Methods, 2012,9: 333-337
- Del Angel V D, Hjerde E, Sterck L, et al. Ten steps to get started in Genome Assembly and Annotation. F1000Research, 2018, 7.


