
 Sample Submission Guidelines
Sample Submission Guidelines
Low-coverage Whole Genome Sequencing
What is low-coverage genome sequencing?
Genotypes detected using high-depth resequencing methods are undoubtedly the most comprehensive, but are currently too costly to apply to plant and animal breeding, especially for species with complex, large genomes. Low-coverage genome sequencing (lcWGS) is emerging as an effective alternative by sacrificing sequencing depth for greater genome coverage and larger sample size with the help of probabilistic statistical strategies.
The LcWGS strategy combines the advantages of RAD-seq, while avoiding the disadvantages. It can study the whole genome at the population level (considering both genome depth and breadth), while retaining the information of individuals, and the cost is comparable to both. Therefore, obtaining genome-wide genotypes by lcWGS combined with algorithms is a popular practice in recent years.
Features of low-coverage whole genome sequencing technology
| LcWGS | WGS | Array | RAD-seq | |
| Sequencing depth | low | high | - | high |
| Number of variants | more | more | less | less |
| New variant detection | yes | yes | no | no |
| Accuracy | moderate | high | high | high |
| Reference genome | yes | yes | yes | yes/no |
| Cost | low | high | low | low |
What problems can LcWGS solve?
Low-coverage genome sequencing (lcWGS) first performs whole-genome low-depth resequencing and variant detection for all individuals in a population, and then uses algorithms to infer and fill (Imputation) the missing genotypes based on the linkage disequilibrium (LD) between variants, and finally obtains high-density genetic markers at the whole-genome level for large-scale samples.
In recent years, LcWGS for large samples has been theoretically demonstrated to obtain genome-wide high-density SNP markers at a very low cost, which in turn increases the accuracy of QTL localization and better explores the genetic mechanisms of various diseases. LcWGS has also been used for association analysis and population genetic studies. The advantage of populating low-density data to the level of whole-genome sequencing for breeding value prediction was found to be highly dependent on the frequency distribution of causal mutations. The superiority of populated data under a neutral model was small, and the accuracy of genetic assessment using populated data could be improved by 30% when all causal mutation minimum allele frequencies were low.
Workflow of LcWGS analysis
The LcWGS pre-processing process is similar to WGS, but an important difference is the need to use genotypic probabilities to explain the probability of genotypic uncertainty, such as downstream analysis using the site frequency spectrum (SFS). lcWGS data analysis process, which uses genotypic probabilities to explain the genotypic A probabilistic framework for uncertainty. From allele frequency spectrum (SFS) to diversity statistics and FST, is the analysis process of ANGSD software. Other tools (e.g. ATLAS) can infer these statistics directly from GLs without prior use of SFS.
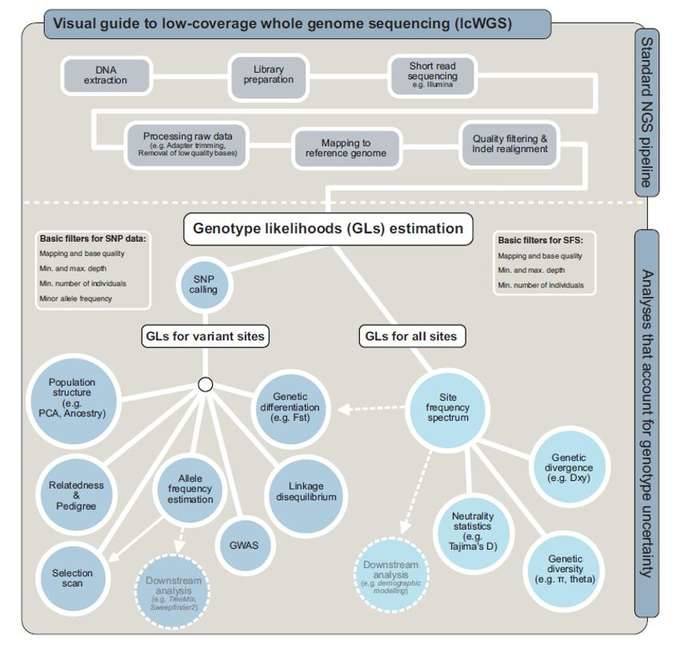 Workflow of lcWGS. (Lou et al., 2021)
Workflow of lcWGS. (Lou et al., 2021)
How to design experiments for low-coverage whole genome sequencing?
There is no single set of experimental designs for low-depth resequencing that is suitable for all study purposes. Instead, the optimal design depends on the study's objectives, system, and budget. Given a budget, the main trade-offs for low-depth resequencing are sample size and sequencing depth. For example, allele frequency estimation, population structure analysis, and genetic differentiation between populations can be sequenced with more samples to obtain accurate results; allele frequency spectra (SFS), demographic inference using δaδi, Tajima'D, or LD absolute values require consideration of higher sequencing depths. Therefore, researchers must carefully consider which type of analysis is most important for the study objectives and find the appropriate balance. Synthesizing the results of our and previous studies, we provide some general guidelines for the design of low-depth resequencing experiments.
What are the advantages of low-coverage genome sequencing?
Low-coverage genome sequencing refers to the sequencing of a genome at a relatively low depth, typically resulting in incomplete coverage of the entire genome. While low-coverage sequencing has some advantages, it also comes with certain limitations.
Advantages of low-coverage genome sequencing:
- Cost-effective: Low-coverage sequencing requires fewer sequencing resources, making it more cost-effective compared to high-depth sequencing. This allows researchers to sequence a larger number of samples within a given budget.
- Efficient for population studies: In population studies, where researchers aim to analyze genetic variations across a large number of individuals, low-coverage sequencing can provide a reasonable representation of genetic variation at a fraction of the cost of deep sequencing. It allows for efficient identification of common genetic variants and the exploration of population-level patterns.
- Identification of common genetic variants: Low-coverage sequencing can effectively identify common genetic variants that occur at a higher frequency in the population. This is particularly useful in genome-wide association studies (GWAS), where researchers investigate the relationship between genetic variations and traits or diseases.
How about the limitations and challenges of low-coverage genome sequencing?
Despite the many advantages of LcWGS, there are still shortcomings in the following areas:
- Complex process and lack of user-friendly software interface and documentation: LcWGS involves several steps, including library preparation, sequencing, and data analysis. The process can be technically challenging, requiring expertise in genomics and bioinformatics. Furthermore, the availability of user-friendly software interfaces and comprehensive documentation for LcWGS analysis may be limited, making it less accessible to researchers who are not well-versed in these techniques.
- Computational demands for phase fixation and padding: To accurately determine the phase of genetic variants (i.e., the arrangement of variants on each chromosome), additional computational resources and algorithms are required. Phase fixation and padding involve computationally intensive processes to resolve haplotype ambiguity and reduce the number of possible haplotype combinations, making it demanding in terms of time and computational power.
- Inconsistent genotype interpretation due to software limitations: The software used for LcWGS analysis may have certain limitations, such as algorithmic biases or errors in genotype calling. This can lead to inconsistencies in genotype interpretation, affecting the accuracy and reliability of the results.
- Not suitable for calling analyses of known genotypes and susceptible to batch effects: LcWGS may not be ideal for calling analyses of known genotypes, such as targeted genotyping or validation studies. The limited coverage and depth may result in missed or inaccurate genotyping calls, impacting the reliability of such analyses. Additionally, LcWGS can be susceptible to batch effects, where technical variations introduced during sample processing or sequencing can influence the results, making it challenging to compare samples across different batches or studies.
- Inability to accurately phase without a reference panel: Accurate phasing of genetic variants requires a reference panel of haplotypes from a diverse population. Without a suitable reference panel, LcWGS may struggle to accurately determine haplotypes, limiting its ability to provide detailed information about the arrangement of variants on each chromosome.
- Unsuitability for small sample sizes and complex genomes: LcWGS may not be well-suited for small sample sizes, as the limited coverage and depth can lead to reduced sensitivity in detecting genetic variants, particularly rare or low-frequency ones. Additionally, complex genomes with repetitive regions or structural variations can pose challenges for accurate variant calling and assembly using LcWGS data.
Reference:
- Lou, Runyang Nicolas, et al. "A beginner's guide to low coverage whole genome sequencing for population genomics." Molecular Ecology 30.23 (2021): 5966-5993.


