
 Sample Submission Guidelines
Sample Submission Guidelines
The Pan-Genome of Arabidopsis thaliana
Introduction
As sequencing technology continues to advance and the quality of genome assemblies continues to improve, more and more high-quality genomes are being released in the same species, providing genetic resources for comprehensive studies of variation in species genomes. A pangenome is a collection of all sequences of a species, including a large number of concordant sequences, large structural variants (SVs) and small variants (such as SNPs, InDels). Simple linear pangenomes cannot visually characterize structural variation, whereas graph-based pangenome assembly can efficiently integrate genetic variation across all newly assembled genomes.
Our Pan-Genome Service
The construction of our pan-genome map not only yields a more complete genetic information of the species, but also allows us to obtain the variation of each individual or population by comparing the sequenced individuals with the reference genome: from simple single nucleotide polymorphisms (SNPs), insertion-deletion mutations (InDel), to large segments of structural variation (SV), copy number variation (CNV), and presence/absence (PAV) variations. In addition, the mechanism of generating shared and unique phenotypes of species can also be explored in depth through comparative analysis of the functions and properties of genes in the core/variable genomes.
We also provide long-read sequencing services to promote pan-genome assembly projects. PacBio HiFi reads are characterized by long read length and high accuracy, which can be used for genome assembly without the need for self-correction of long-read sequencing data and correction of NGS data, thus effectively saving the analysis time and computational resources, and can be used for pan-genome research after assembly of low-depth (10-15X) HiFi reads. Compared with the high-depth next-generation sequencing data, HiFi reads can be assembled with better effect, and the pan-genome information obtained is more accurate, which can be used for the detection of all variants (SNP, InDel, SV, CNV, PAV), and avoids the drawbacks of the NGS data, such as the short read length.
High-Quality Genome Assembly of A. thaliana
This study completed a high-quality genome assembly of 32 representative A. thaliana ecotypes using the PacBio HiFi sequencing strategy, of which 6 ecotypes are solanaceous ecotypes with distinct differences, and the other 26 ecotypes are non-solanaceous ecotypes that underwent monophyletic expansion after the ice age. Pan-genomic analysis based on gene family clustering identified variable genes in Arabidopsis that expanded the gene pool of different ecotypes and thus contributed to adaptation to the local environment. To identify structural variants (SVs) in the 32 ecotypes, the researchers further constructed a graphic pangenome and identified 61,332 large-band structural variants (>50 bp) based on the graphic pangenome, covering 18,883 genes, some of which are highly correlated with ecological adaptations in Arabidopsis. For example, a specific 332 bp insertion in the promoter region of the HPCA1 gene in the Tibetan ecotype (Tibet-0) resulted in up-regulated expression of the gene, which facilitates Arabidopsis adaptation to alpine environments. This study reveals the genetic contribution of structural variation to local adaptation in different ecotypes of Arabidopsis.
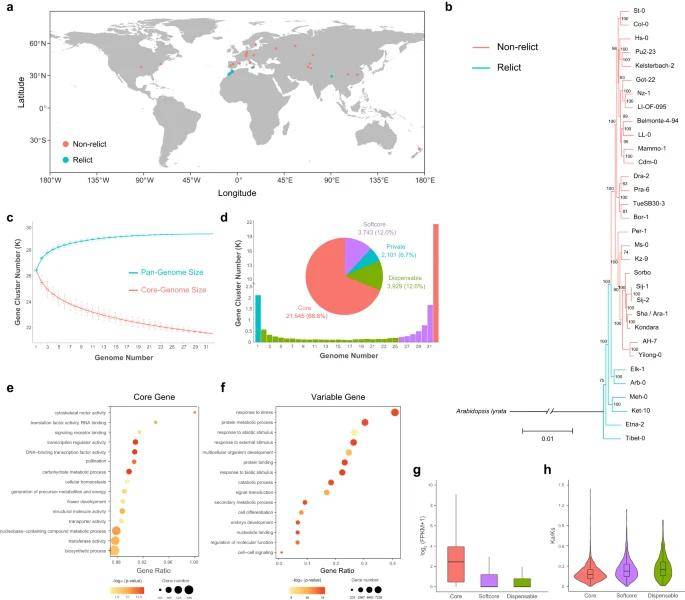 Pan-genome of 32 A. thaliana ecotypes. (Kang et al., 2023)
Pan-genome of 32 A. thaliana ecotypes. (Kang et al., 2023)
Reference:
- Kang, Minghui, et al. "The pan-genome and local adaptation of Arabidopsis thaliana." Nature Communications 14.1 (2023): 6259.


