
 Sample Submission Guidelines
Sample Submission Guidelines
The Human Gut Virome Unveils a Multitude of dsDNA Phage Genomes
dsDNA Phages
This rich community of bacteriophages (phages) exerts a profound influence on the structure and function of the bacteriome, primarily through bacterial lysis and gene transfer. However, the impact of population-scale virome mutations remains largely unexplored.
The enteroviral group, dominated by dsDNA phages, rivals bacterial populations in numbers within the human gut. Notable members of these dsDNA phages include crAssphage, Lak phage, and Gubaphage/Flandersviridae. Their genomes offer valuable insights into their functional potential, unique biological characteristics, and ecological roles within the human gut. Extensive research has been conducted on these phage genomes, and they are now accessible in multiple languages.
In a comprehensive study, they collected 4,198 fecal samples from Japanese individuals (averaging 66.4 ± 12.6 years in age) participating in the 4D program. These samples underwent metagenome sequencing, and meticulous data gathering encompassed intrinsic and extrinsic factors such as age, gender, body mass index, lifestyle, dietary habits, diseases, medications, and more. Subsequently, they employed an innovative analytical process to establish a high-quality phage catalog and conducted in-depth analyses.
The investigation of human enteroviruses derived from the intestinal metagenomes of these 4,198 individuals unveiled a plethora of high-quality dsDNA phage genomes. Furthermore, through comprehensive correlation analyses involving various host and environmental factors, we identified numerous intrinsic and extrinsic elements significantly associated with viral group structure.
Establishing a Robust Phage Genome Catalog in a Japanese Microbiome Cohort
In their study, a novel approach was introduced to construct a comprehensive phage genome catalog within a microbiome cohort in Japan. This innovative process leverages the identification of phage genome-specific features, setting it apart from existing viral detection tools. Their findings indicate that while the true-positive rate is on par with or slightly lower than alternative methods, the false-positive rate is significantly reduced, standing at just 0.4%. These results suggest that their approach is well-suited for creating high-quality phage catalogs with minimal non-phage sequence contamination.
Through their efforts, they successfully identified a total of 1,125 complete phage genomes and 3,584 sketched phage genomes (comprising over 70% completeness) within a cohort of 4,198 Japanese individuals. A remarkable 59.9% of these genomes exhibited high quality. To assess the coverage of this catalog in relation to double-stranded DNA (dsDNA) phages in the human gut, virus-like particle (VLP) sequencing was performed on an additional 24 fecal samples. Their results were found to be in alignment with another phage genome catalog generated using the VIBRANT software.
They employed host prediction techniques based on the CRISPR spacer region, which revealed the most prevalent phages belonged to the Thickettsia, Anaplasma, and Actinobacteria phyla, and at the genus level, their common hosts included Anaplasma, Ruminalococcus, Blautia, and Bifidobacterium. A noteworthy observation was that a significant portion of phages (71.1%) exclusively infected a single genus, designating them as endemic phages, while others exhibited a broader host range, capable of infecting multiple genera, thus categorized as universal phages. The sizes of phage genomes within the Mycobacterium, and Prevotella phage phylum were relatively large, consistent with variations in host genome size.
Additionally, clustering of viral Operational Taxonomic Units (vOTUs) based on the proportion of shared proteins exceeding 20% led to the identification of a total of 223 clusters representing virus families (VCs) or subfamilies.
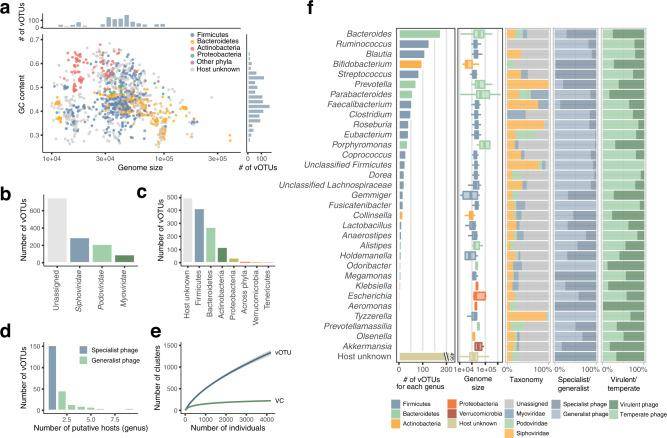 Overview of reconstructed phage genomes from 4198 human gut metagenomes. (Nishijimaet al., 2022)
Overview of reconstructed phage genomes from 4198 human gut metagenomes. (Nishijimaet al., 2022)
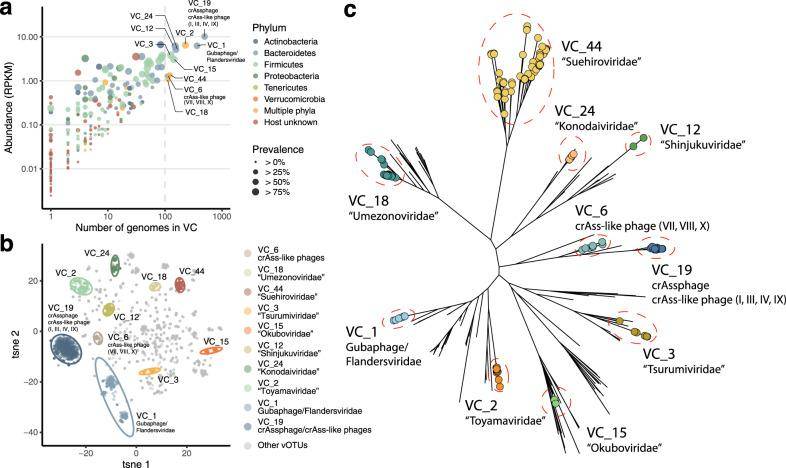 Identification of novel viral clusters abundant and prevalent in the human gut. (Nishijimaet al., 2022)
Identification of novel viral clusters abundant and prevalent in the human gut. (Nishijimaet al., 2022)
Intimate Relationships Between Gut Viruses and Bacteria
When comparing the characteristics of viruses and bacteria collected from 4,198 individuals, a profound connection emerged. There was a significant positive correlation between their α and β diversity, unveiling a close structural relationship between viral and bacterial compositions in the human gut. Interestingly, it was noted that viruses exhibited a considerably higher β diversity compared to bacteria, implying that viruses possess a more individual-specific nature than bacteria.
Intriguingly, one-to-one correlations were discovered between the relative abundance of each phage and its predicted host at the genus level within the 4,198 individuals. This finding suggests that phages and their host bacteria engage in a mutually beneficial symbiosis within the human gut. Furthermore, it was observed that the correlation between endemic phages and their hosts was significantly stronger compared to universal phages, shedding light on the specificity of these interactions.
To delve deeper into these relationships, an exploration of antiviral genes, including CRISPR-Cas systems, was conducted. The abundance of these genes was quantified, and their correlation with the virological structure was examined. The results uncovered a significant increase in the Shannon index of viruses in samples with high abundance of defense genes, across all three prokaryotic defense systems, when compared to samples with low abundance of these defense genes.
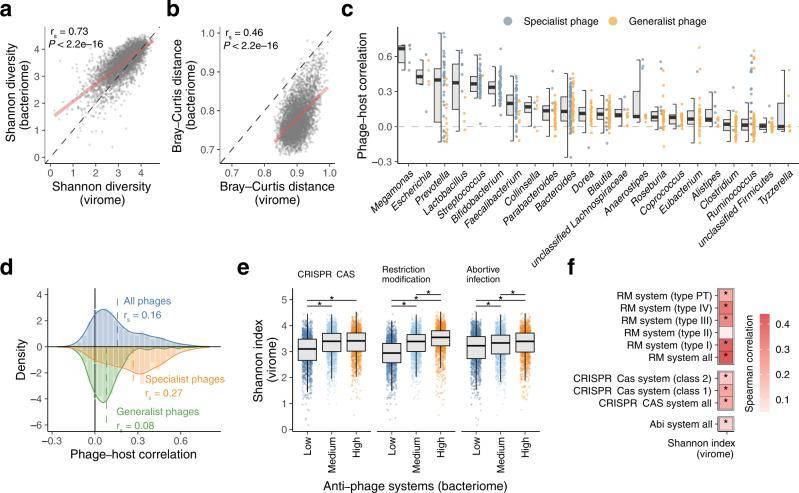 Close interactions between the gut virome and bacteriome. (Nishijimaet al., 2022)
Close interactions between the gut virome and bacteriome. (Nishijimaet al., 2022)
Comprehensive Analysis of Host and Environmental Factors Impacting Virology
In their exploration of the relationship between the structure of enteroviruses and the physiological and environmental aspects of their hosts, a series of intriguing findings emerged. Age proved to be a pivotal factor, demonstrating a significant and positive correlation with viral diversity, along with a positive association with bacterial diversity. Notably, they found that age exhibited strong connections with 176 viral Operational Taxonomic Units (vOTUs), showing positive associations with Clostridium difficile, Clostridium tumefaciens, Bacillus faecalis phages, and host-unknown phages. On the flip side, age displayed an inverse correlation with VC_28. Similarly, sex displayed significant correlations with 68 vOTUs and 24 virus clusters (VCs).
Expanding their analysis, they evaluated 232 host and environmental factors to investigate their relationships with viral characteristics. The most robust associations, influencing enteroviral diversity, were observed with clinical factors, specifically medications and diseases. Ninety-seven out of the 232 factors displayed significant correlations with viral variation, with age standing out as the factor with the strongest correlation to virus dynamics.
A particularly intriguing discovery was made regarding crAssphage (vOTU_974). It exhibited no significant correlations with any of the analyzed factors, indicating the presence of as-yet-unknown host, environmental, or ecological factors contributing to the variations observed in this exceedingly abundant phage.
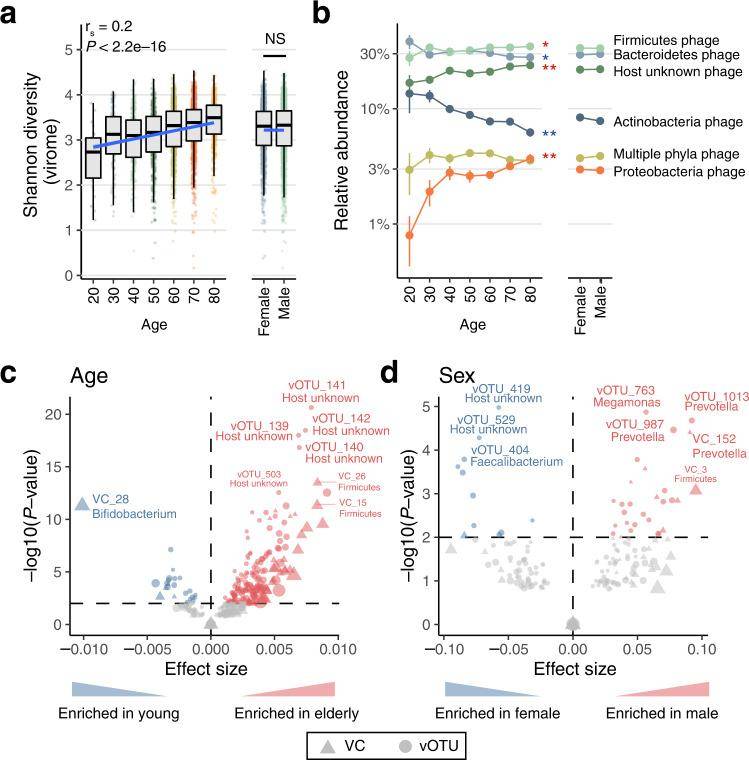 Age- and sex-related changes in the human gut virome. (Nishijimaet al., 2022)
Age- and sex-related changes in the human gut virome. (Nishijimaet al., 2022)
Reference:
- Nishijima, Suguru, et al. "Extensive gut virome variation and its associations with host and environmental factors in a population-level cohort." Nature Communications 13.1 (2022): 5252.


