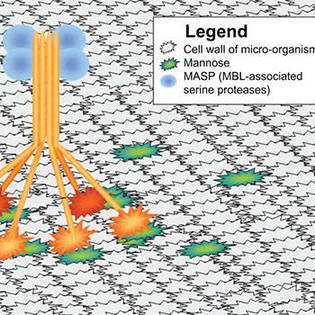
The complement system is a rapid and efficient immune surveillance system and coordinates innate and acquired immunity by augmenting antibody (Ab)responses and immunologic memory, lysing foreign cells, and clearing immune complexes and apoptotic cells. The system contains plentiful soluble proteins found in the circulation and in tissues. The system, as an enzyme cascade, helps host eliminate cellular debris and infectious microbes, orchestrates immune responses, and informs the body of invading signals through three activation pathways: the classical, alternative and lectin pathway. On one hand, the complement system contributes to homeostasis; on the other hand, the system will attack healthy cells if under improper control. The complement system disorder represents either deficiencies or inappropriate activation. The deficiency in any component of the system can result in immunocompromise, overwhelming infection and sepsis. At the same time, uncontrolled complement activity can also cause disease through promoting inflammation. Studies have found disorders of complement system is involved in various types of diseases, including Paroxysmal Nocturnal Hemoglobinuria (PNH), atypical Hemolytic Uremic Syndrome (aHUS), Neuromyelitis Optica (NMO), hereditary angioedema, neurodegenerative disorders, type I diabetes mellitus, cancers, Alzheimer’s disease, and so forth. Cause of the complement system disorders can be primary or secondary in nature, and congenital complement deficiencies have been reported in all the individual components of complement and most of the regulatory components.
The activation of complement system depends on three pathways, and mutations in any of the component of the pathway will lead to disorders. C4A and C4B encode C4, which participates in classical pathway. Deletion of the C4A gene is the most common mechanism, and deficiency of C4A and C4B is related to the development of diabetes mellitus, chronic hepatitis, etc. Another component of classical pathway, C2, is the most common inherited complement deficiency. Homozygous deficiency of C2 will lead to failure in formation of multi protein complex C4b2a that serves as the classical pathway C3 converts activating enzyme. Besides, a somatic mutation of PIGA (Phosphatidylinositol Glycan Anchor Biosynthesis Class A) in the hematopoietic stem cells causes dysfunction of glycosyl-phosphatidylinositol molecule, which anchors about 20 proteins to the cell membrane, including Decay Accelerating factor (DAF) and CD59. Consequently, the absence of these protein makes the erythrocytes more susceptible to complement-mediated lysis. In addition, heterozygous mutations in the membrane cofactor protein gene CD46 has been found in aHUS, Stx-HUS and the HELLP (hemolysis, elevated liver enzymes, and low platelets) syndrome. In whole, the complement system contains more than 30 serum and membrane-bound proteins. Any mutations in genes encoding complement receptors, binding proteins, etc., will result in malfunction of complement system.
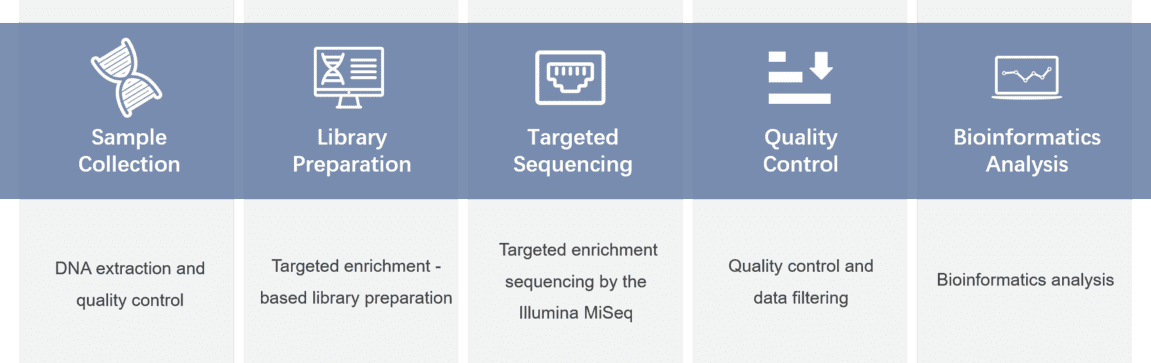
To understand the gene variants in the complement system disorders better, targeted enrichment technology by Illumina MiSeq system will be performed in our platform. We offer a comprehensive complement system disorder panel for your research in the complement system disorder. You can choose genes for interest from our panel or discuss about your own designed panel.
| ADIPOQ | ADIPOR1 | ADIPOR2 | ARMC4 | C1QA | C1QB | C1QC | C1R |
| C1S | C2 | C3 | C3AR1 | C4A | C4B | C4BPA | C4BPB |
| C5 | C5AR1 | C5AR2 | C6 | C7 | C8A | C8B | C8G |
| C9 | CCDC114 | CCDC39 | CCDC40 | CCNO | CD46 | CD55 | CD59 |
| CD93 | CD93 | CF1 | CFB | CFD | CFH | CFHR1 | CFHR2 |
| CFHR3 | CFHR4 | CFHR5 | CFI | CFP | CLU | COLEC11 | CR1 |
| CR2 | CRP | CSMD1 | CSMD2 | CSMD3 | DGKE | DNAAF1 | DNAAF2 |
| DNAAF3 | DNAAF5 | DNAH11 | DNAH5 | DNAI1 | DNAI2 | DNAL1 | DRC1 |
| DYX1C1 | FCN1 | FCN2 | FCN3 | FCN3 | HYDIN | ITGAL | ITGAM |
| ITGAX | ITGB2 | LRRC6 | MASP1 | MASP2 | MASP2 | MBL2 | NME8 |
| OFD1 | PIGA | PTX3 | RSPH1 | RSPH4A | RSPH9 | SERPING1 | SPAG1 |
| THBD | VSIG4 | VTN | ZMYND10 |
For more information about the Custom Complement System Disorders Panel or need other amplification requirements, please contact us.
References:
Please submit a detailed description of your project. We will provide you with a customized project plan to meet your research requests. You can also send emails directly to for inquiries.
Please fill out the form below: ×