Fatty acid oxidation disorder is a kind of disease caused by deficiency of enzyme needed for oxidize fatty acid. Fatty acids in the body are divided into short chains (2-4 carbon), medium chains (4-12 carbon) and long chains (more than 12 carbon), depending on how many C atoms they contain. There are two kinds of organs in human body to complete the oxidation metabolism of fatty acid β: mitochondria and peroxidation objects. Mitochondria are mainly engaged in the metabolism of fatty acid with ≤20 carbon units. So far, at least 25 enzymes or transporters involved in the oxidation of fatty acid beta have been found. Abnormalities in any of the fatty acid oxidation links can lead to the disturbance of fatty acid decomposition and energy generation, and the dysfunction of nervous system, skeletal muscle, heart, liver, kidney, digestive tract, etc.
The obstacles in the process of fatty acid metabolism include: fatty acid and carnitine transport disorders, CoA dehydrogenase deficiency, dysfunction of various enzymes required for β-oxidation in the mitochondrial matrix, the disorder of ketone body formation or the combination of several defects exist. In addition, pregnancy can also induce abnormal beta-oxidation metabolism. All the metabolic defects limited to mitochondrial fatty acid metabolism disorders are autosomal recessive with great genetic heterogeneity. Fatty acid and carnitine transport disorders include cellular uptake of fatty acid barriers, cytosolic carnitine transport disorders (primary carnitine defects), CPT I deficiency, carnitine-CAT deficiency and CPT II deficiency. CPT II deficiency is the most common type of fatty acid metabolism disorder. Defects in acyl-CoA dehydrogenase (ACD) include very long-chain, medium-chain, and short-chain. 80% of medium-chain ACD (MACD) defects in the amino acid residue 985 locus adenine are replaced by guanine (985 A>G). Short-chain ACD (SACD) defects often have two mutations, namely 625G>A and 511C>T. The mutations in ETF gene are present in individuals with multiple CoA dehydrogenase abnormalities (MADD). Mutations in the HADH and HADHA genes result in defects in long-chain and short-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD, SCHAD), respectively. More genes are listed in fatty acid oxidation disorder gene list.
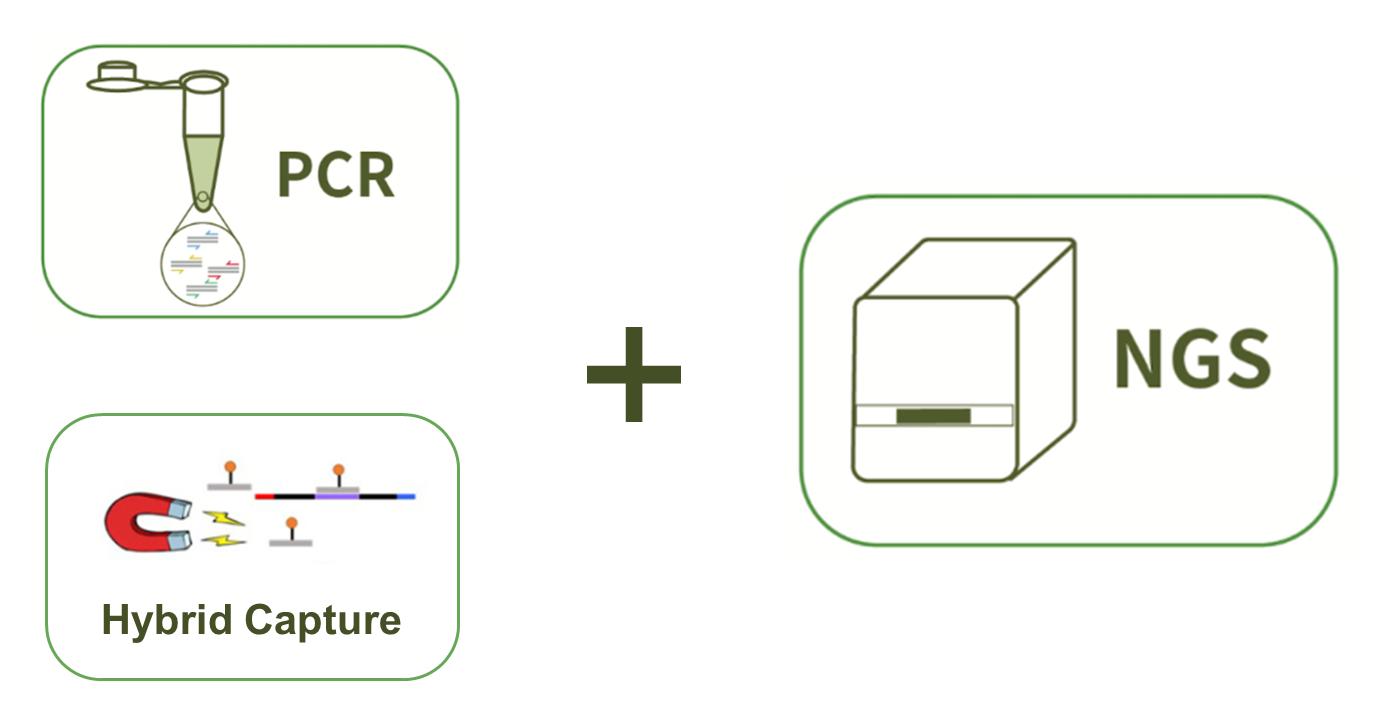
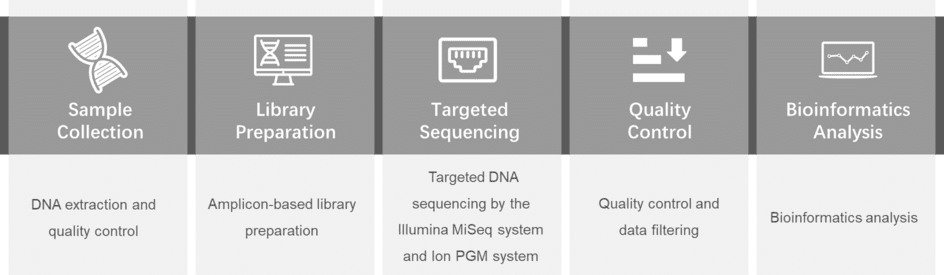
To better understand and support for the study of genetic alterations in fatty acid oxidation disorder, CD-Genomics provides a custom fatty acid oxidation disorder panel containing optimized genes which are reported associated with the increase of risk of fatty acid oxidation disorder. Targeted DNA sequencing technology by Illumina MiSeq system is provided to detect the genes you are interested in.
| ACAD8 | ACAD9 | ACADM |
| ACADS | ACADVL | CPT1A |
| CPT2 | ETFA | ETFB |
| ETFDH | GLUD1 | HADH |
| HADHA | HADHB | HMGCL |
| HMGCS2 | HSD17B10 | LPIN1 |
| MLYCD | PPARG | SLC22A5 |
| SLC25A20 | TAZ |
For more information about the Custom Fatty Acid Oxidation Disorder Panel or need other amplification requirements, please contact us.
References:
Please submit a detailed description of your project. We will provide you with a customized project plan to meet your research requests. You can also send emails directly to for inquiries.
Please fill out the form below: ×