Cystic fibrosis is an autosomal recessive congenital disease, caused by the mutation of cystic fibrosis transmembrane conductance regulator (CFTR) which is a cAMP-dependent chloride channel protein belongs to the ATP binding cassette transport proteins superfamily. CFTR distributes in the tissue epithelial cells which are related to secretion and absorption. Dysfunction of CFTR protein will destroy the channel on the surface which allows the chloride ion and sodium ion get in and out of the cells. The reduction of airway surface liquid impedes mucosal cilia swing and mucus removal. Clinical manifestations of cystic fibrosis include chronic airway mucus accumulation and microbial growth. Inflammatory cell invasion into the lungs causes the persistent inflammatory response, this response leads to impaired lung function and more severe symptoms.
CFTR gene, maps to 7th autosome (7q31-7q32), including 27 exons and 26 introns and is highly polymorphic. Currently it is reported that CFTR gene has 1000 mutant forms and 700 positive mutant forms, which are divided into 6 categories. Class I includes frameshift, splicing and nonsense mutations, which result in severe reducing or deletion of CFTR expression. W1282X is a representative class I mutant type. The mutations of class II lead to misfolding and premature degradation of the endoplasmic reticulum (ER) quality-control system, severely decreasing the mature CFTR molecules that reach the cell surface. △F508 and P67L are mutations with different degrees of class II, and the former is more serious. Class III mutations impair the regulation of the CFTR channel, resulting in abnormal chloride channel function. Class IV mutations influence the ion conduction to alter the channel conductance, but the distinguish of these two extents is not clear. R117H, G551D and S1251N mutant types belong to class III/IV. Class V mutations impede the abundance of the CFTR by introducing promoter or splicing abnormalities. Class VI mutations lead to the decreasing stability of CFTR protein, resulting in the out control of ion channel. Because of the diversity of mutations, regulation mechanism of CF disease is a complex network.
Our platform provides amplicon sequencing technology with the Illumina MiSeq or Ion PGM system to research the mutations of CFTR and the mechanism of cystic fibrosis. We also offer a comprehensive cystic fibrosis panel library for you to choose for genetic testing of cystic fibrosis.
| CFTR |
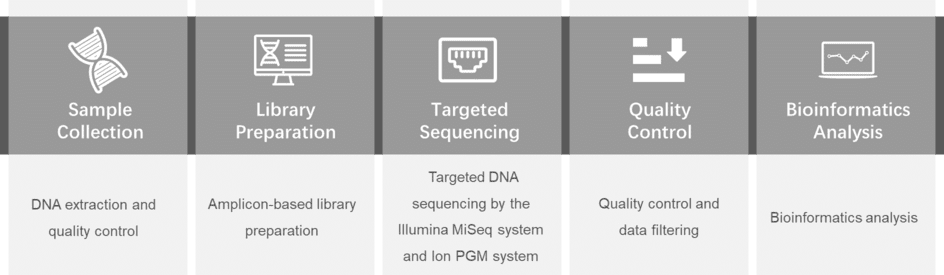
For more information about the Custom Cystic Fibrosis Panel or need other amplification requirements, please contact us.
References:
Please submit a detailed description of your project. We will provide you with a customized project plan to meet your research requests. You can also send emails directly to for inquiries.
Please fill out the form below: ×