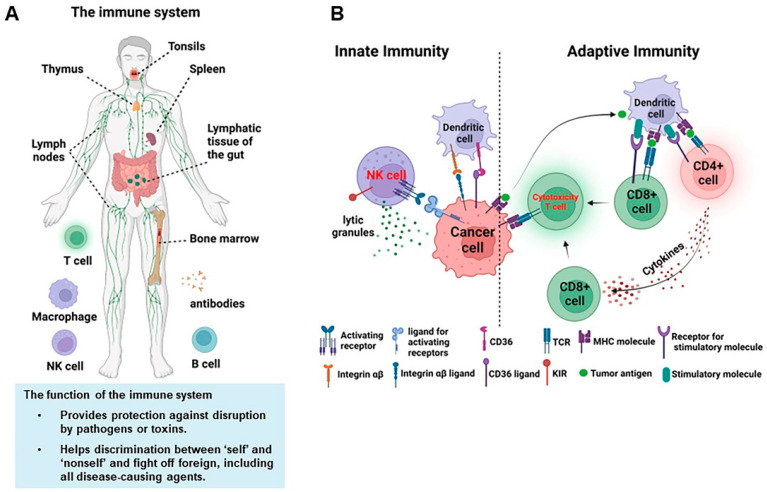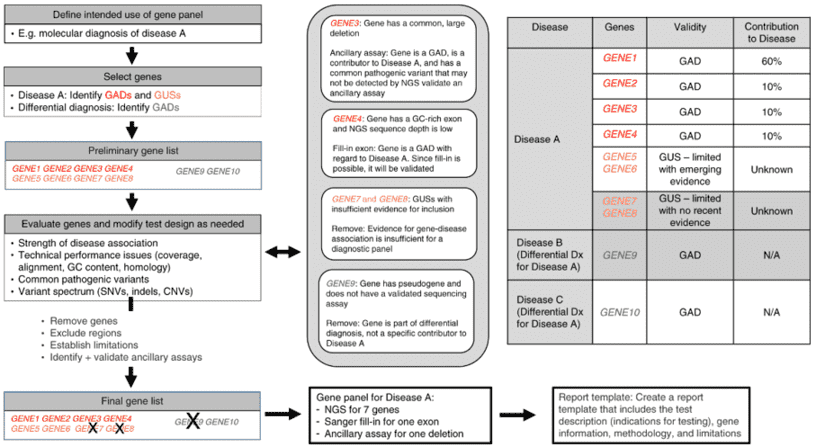
Lysosomal storage disorders (LSDs) are hereditary metabolic diseases caused by the deficiency of enzymes, activators, transporters or lysosomal protein processing and correcting enzymes in the lysosome, resulting in lysosomal functional defects and the inability to digest metabolites effectively, thus resulting in storage in tissues and cells. Most LSDs are autosomal recessive and a few are X-linked recessive. In general, LSDs is one of the most common genetic diseases in humans, the incidence of one of the LSDs is very low. According to the defective proteins or storage species, LSDs are divided into 7 categories: glycosaminoglycan metabolism defects, glycogen decomposition defects, nerve sphingolipid decomposition defects, cholesterol and cholesterol ester decomposition defects, various lysosomal enzyme defects, transport and transport defects.
Mucopolysaccharide storage disease, mucolipid storage disease, sphingolipid deposition disease and glycogen storage disease, especially glycogen storage disease type II, are common types in LSD. The genetic factors leading to mucopolysaccharide disease are a series of lysosomal hydrolase defects related to mucopolysaccharide degradation. The pathogenic mutations in IDUA, IDS, GALNS and GLB1 genes are related to the molecular genetics of type I, II, IVA and IVB mucopolysaccharidosis, respectively. Shear mutations and nonsense mutations in the GNPTAB gene are key factors in the development of mucolipidosis. Neuronal ceroid lipofuscinose (NCL) is a group of lysosomal lipofuscinosis disease. In polytype NCLs, there are biochemical abnormalities in lysosomal hydrolase in cells and significant deposition of subunit C of mitochondrial ATP synthase. Mutations in at least 8 different genes (CLN1~8) have been found to cause NCLs. Mutation of the glucocerebrosidase GBA gene causes a decrease or loss of glucocerebrosidase activity, letting glucocerebroside to accumulate in mononuclear-macrophages, and leading to Gaucher disease. Lysosomal storage diseases contain a wide variety of species, and more genes associated with these diseases can be viewed in the gene list. Because of polymorphic genetic variation, some mutations in the ARSA, HEXA, GAA, GLA, GLB1, FUCA1 and GUSB genes will result in greatly reduced enzyme activity, while the individual remains clinically healthy, a phenomenon known as enzyme pseudodeficiency (Pd).
To support researches related to lysosomal storage disorders associated genes, our custom lysosomal storage disorders panel platform offers a comprehensive lysosomal storage disorders panel library from which you can choose for genetic testing of LSDs. High-throughput targeted enrichment sequencing technology that we perform can screen genetic variants among the lysosomal storage disorders gene panel via a high effective mode and help researchers decode the biology of LSDs.
| ABCD1 | ABHD5 | ACOX1 | ADAMTSL2 | AGA | AGPS | AGXT | ANTXR2 |
| ARSA | ARSB | ASAH1 | ATP6AP1 | CLN3 | CLN4 | CLN5 | CLN6 |
| CLN8 | CTNS | CTSA | CTSC | CTSD | CTSF | CTSK | DHCR7 |
| DNAJC5 | DNM1L | DYM | FUCA1 | GAA | GALC | GALNS | GBA |
| GLA | GLB1 | GM2A | GNE | GNPAT | GNPTAB | GNPTG | GNS |
| GPC3 | GUSB | HEXA | HEXB | HGSNAT | HRAS | HYAL1 | IDS |
| IDUA | KCTD7 | LAMP2 | LIPA | MAN2B1 | MANBA | MCOLN1 | MFSD8 |
| NAGA | NAGLU | NEU1 | NPC1 | NPC2 | PEX1 | PEX10 | PEX12 |
| PEX13 | PEX14 | PEX16 | PEX19 | PEX2 | PEX26 | PEX3 | PEX5 |
| PEX6 | PEX7 | PHYH | PNPLA2 | PPT1 | PSAP | RAI1 | SCARB2 |
| SGSH | SLC17A5 | SMPD1 | SUMF1 | TCF4 | TPP1 | TRIM37 | VPS33A |
For more information about the Custom Lysosomal Storage Disorders Panel or need other amplification requirements, please contact us.
References:
Please submit a detailed description of your project. We will provide you with a customized project plan to meet your research requests. You can also send emails directly to for inquiries.
Please fill out the form below: ×